超聲-微波協同提取大青葉多糖及其分子特性和免疫活性研究
2019-03-08 08:31:06曹榮安李朝陽李昌盛張開江王長遠
天然產物研究與開發 2019年2期
曹榮安,李朝陽,李昌盛,張開江,王長遠
1黑龍江八一農墾大學食品學院;2黑龍江省農產品加工與質量安全重點實驗室;3黑龍江八一農墾大學國家雜糧工程技術研究中心,大慶 163319;4韓國江陵原州大學海洋食品科學技術系,江陵 210702;5大慶市燃乏中藥材種植專業合作社,大慶 163411
大青葉是十字花科(Cruciferae)菘藍屬(Isatis)植物菘藍(IsatisindigoticaFort)的干燥葉,其地下部分為板藍根,在我國中東部分布較廣[1]。大青葉是我國傳統中藥,性寒、味苦、歸心胃經[2]。經研究發現,大青葉中存在有機酸、靛藍、靛玉紅、菘藍苷、色胺酮、喹唑酮、異牡荊素、多糖等化學成分化合物[3]。現代藥理研究表明,大青葉具有清除自由基、調理血脂、抗病毒、抗菌、抗癌、增強免疫調節等作用[4,5],具有清熱解毒、涼血消斑之功效,臨床上可用于溫病高熱、神昏、發斑發疹、作腮、喉痹、丹毒、癰腫[6]。目前對于大青葉中化學成分的研究主要集中在小分子物質,對多糖的研究報道很少[7]。本文是以大慶地產大青葉為原料,利用超聲-微波協同水提法進行大青葉多糖提取,研究提取參數,分析大青葉多糖化學成分、單糖組成、分子量和鏈接方式,同時進行體外生物活性研究,為大青葉多糖的研究利用提供理論基礎。
1 材料與方法
1.1 材料與儀器
大青葉由大慶市燃乏中藥材種植專業合作社提供,RAW264.7細胞來自ATCC。RPMI-1640細胞培養基、青霉素/鏈霉素、牛血清蛋白購自美國Lonza公司,脂多糖(LPS)、Griess (modified)購自美國Sigma-Aldrich公司,EZ-cytox細胞增殖及細胞毒性檢測試劑盒(2-(4-碘苯)-3-(4-硝基苯)-5-(2,4-二磺基苯)-2H-四氮唑鈉鹽,WST-1)購自韓國Daeillab公司,蛋白質定量試劑盒購自美國Bio-Rad公司。其他化學試劑均為分析純。
Excella ECO-170細胞培養箱,英國New Brunswick Scientific公司;EL800酶標儀,美國Biotech公司;6890N/MSD5973氣質聯用儀(GC-MS),美國Agilent公司;LR64912C傅里葉近紅外光譜儀,美國PerkinElmer;高效尺寸排阻色譜-多角度激光光散射儀-示差折光檢測器聯用系統(HPSEC-MALLS-RI),包括泵(#321,Gilson,Middleton,WI,USA),注射器(#7072,Rheodyne),尺寸排阻色譜柱(TSK G5000 PW;7.5×600 mm,TosoBiosep,Mongomeryville,PA,USA),MALLS檢測器(HELEOS;Wyatt Technology Corp,Santa Barbara,CA,USA),RI檢測器(#2414,Waters);CW-2000超聲-微波協同萃取/反應儀,上海新拓分析儀器科技有限公司。
1.2 方法
1.2.1 大青葉多糖提取
大青葉原料烘干研磨后加入85%乙醇,70 ℃加熱回流攪拌2 h,冷卻后室溫攪拌12 h,離心后殘渣中分別加入85%乙醇和無水丙酮室溫攪拌,自然干燥得到脫脂大青葉。加入蒸餾水在水浴鍋中提取一定時間,之后再放入超聲-微波協同萃取/反應儀中浸提(超聲功率50 W),合計提取時間2 h,離心得到上清液,浸提2次,上清液旋轉蒸發濃縮,加入乙醇使最終乙醇體積分數為80%,攪拌10 min后放入4 ℃冰箱中靜置12 h。離心棄去上清液,沉淀分別加入無水乙醇和丙酮洗滌2次,離心后沉淀室溫干燥。干燥提取物利用Sevag試劑(氯仿與正丁醇體積比為5∶1)去除蛋白質,3 500 D膜透析凍干得到大青葉多糖,稱質量后計算多糖得率,大青葉多糖得率(%)=(提取得到的多糖質量/脫脂后原料質量)×100%。
1.2.2 提取大青葉多糖單因素實驗
1.2.3 提取大青葉多糖正交實驗
根據單因素試驗結果,4個因素各選取3個水平進行正交試驗,從而確定提取大青葉多糖的最佳參數。
1.2.4 化學成分分析
采用苯酚-硫酸法[8]測定總糖含量,采用Lowry法(福林-酚法)[9]測定蛋白質含量,采用氯化鋇-明膠濁度法[10]測定硫酸根含量,采用間羥基聯苯法[11,12]測定糖醛酸含量。
1.2.5 單糖組成和鏈接方式分析
參考文獻[13]方法并稍作改動,樣品經過不同方法的衍生化后注入1 μL到GC-MS儀中進行分析,根據GC出峰時間和MS的離子峰同單糖標準品以及不同鏈接方式的多糖進行對比,從而確定單糖組成和鏈接方式。
在運用該系統的過程最后,車輛進場前,需在項目部安全環保部門辦理臺賬登記手續(三證齊全),手續齊全后發項目部自編號,由設備物資科進行電子標簽的錄入信息工作,錄入完成后裝料,裝料到達自動稱重系統后看指示燈(紅綠燈),當綠燈亮時,車輛行駛到電子標簽掃描區域,掃描成功后道閘打開,車輛進入稱臺中間(此時紅外線已掃描)不用停車方可稱重記錄,記錄成功后語音提示稱重保存成功,顯示屏顯示稱重數據,道閘打開車輛通過稱重完畢。
1.2.6 分子質量及分布測定
參考文獻方法[14],將多糖配置成溶液溶解后利用HPSEC-MALLS-RI聯機系統測定分子質量及分布情況。
1.2.7 紅外光譜分析
取1 mg多糖樣品,按照1∶200(W∶W)的比例與KBr混合,研磨后壓片,利用傅里葉紅外光譜儀掃描分析,掃描范圍4 000~400 cm-1。
1.2.8 多糖對巨噬細胞增殖能力影響
細胞增殖采用WST-1細胞增殖及細胞毒性檢測試劑盒,參考文獻方法[15],RAW264.7細胞用不同濃度的多糖溶液處理后加入WST-1溶液,在450 nm下測定吸光值,計算細胞增殖率。
1.2.9 多糖激活巨噬細胞產生NO測定
參考文獻方法[15],RAW264.7細胞在96微孔板中預培養24 h,細胞中加入多糖溶液,1 μg/mL LPS作為陽性對照,培養24 h。上清液中NO含量按照文獻[16]方法進行測定。
1.3 數據分析
實驗重復3次,數據采用M±SD表示,采用Sigmaplot 12.0作圖。應用SAS 8.2軟件對數據進行統計分析,各實驗組之間的顯著性差異分析采用單因素方差分析(one-way ANOVA)和鄧肯氏復極差法。
2 結果與分析
2.1 大青葉多糖提取參數
2.1.1 單因素結果與分析
影響大青葉多糖得率的4個因素的單因素試驗結果見圖1,其中曲線a是液料比對多糖得率影響,可知在液料比10∶1~25∶1(V/m)范圍內,隨著液料比的增大多糖得率呈顯著上升趨勢(P<0.05),之后隨著液料比升高多糖得率呈現下降趨勢,所以最佳液料比(V/m)范圍為20∶1、25∶1、30∶1。由曲線b可知,在提取溫度75~90 ℃范圍內,隨著溫度升高多糖得率逐漸升高,當提取溫度升高到95 ℃時多糖得率呈現下降趨勢,最佳提取溫度范圍為85、90、95 ℃。從超聲-微波時間對于大青葉多糖得率影響可知(圖1曲線c),當超聲-微波時間在20~50 min時多糖得率一直呈上升趨勢,最佳超聲-微波時間范圍為40、50、60 min進。圖1中曲線d是微波功率對于多糖得率的影響,在微波功率300~500 W內多糖得率呈上升趨勢,之后隨著微波功率升高,多糖得率呈現下降趨勢,微波功率最佳范圍為400、500、600 W。
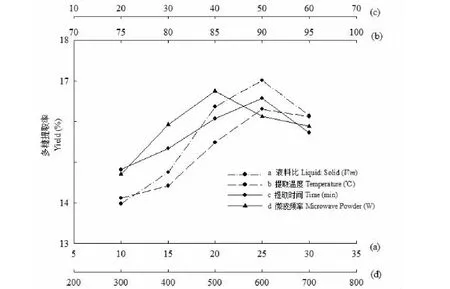
圖1 單因素試驗結果Fig.1 The results of single factor experiment
2.1.2 正交試驗結果
根據單因素試驗結果,選取液料比(V/m)20∶1、25∶1、30∶1,提取溫度85、90、95 ℃,超聲-微波時間40、50、60 min,微波功率400、500、600 W進行正交試驗,試驗設計和結果見表1。
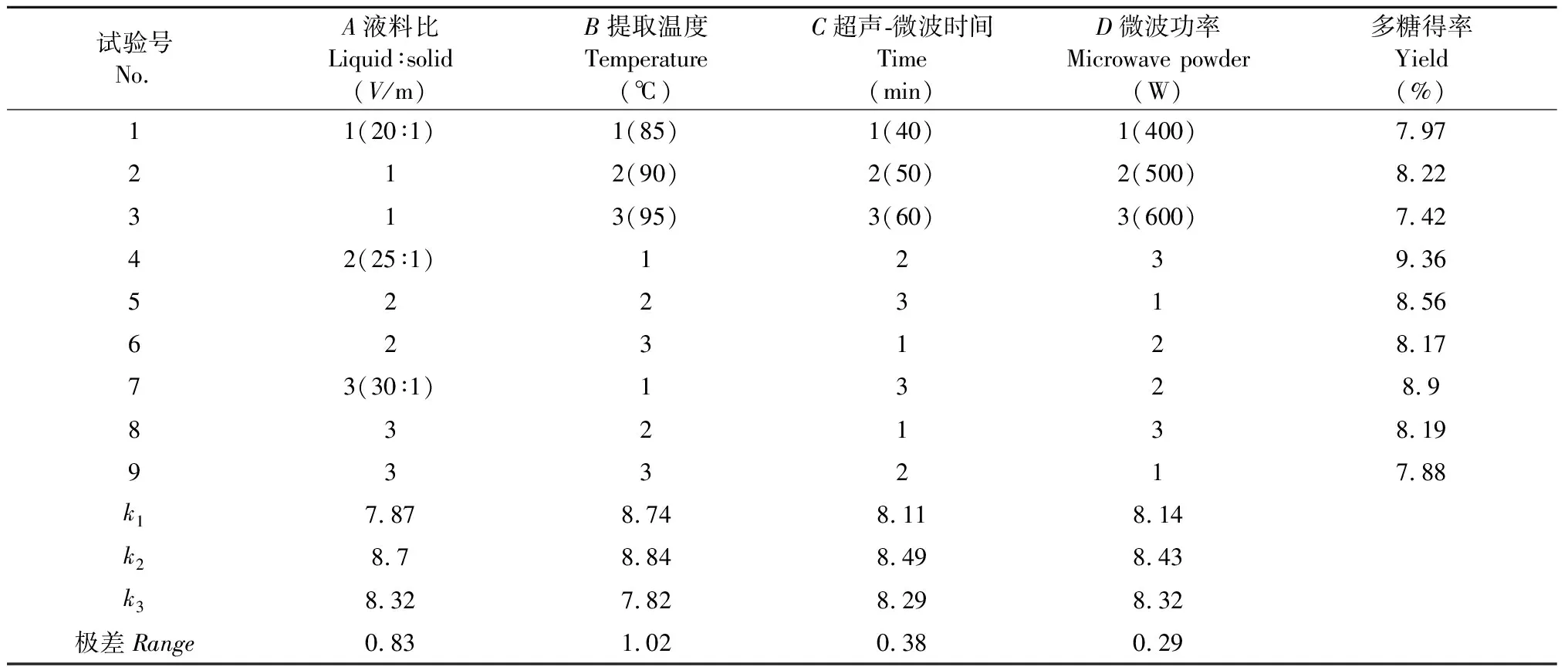
表1 正交試驗設計和結果
根據表1極差分析結果,判斷出各因素對多糖得率影響的主次順序為:B(提取溫度)>A(液料比)>C(超聲-微波時間)>D(微波功率),因此超聲-微波協同提取大青葉多糖的最佳條件是A2B2C2D2,即料液比25∶1,溫度90 ℃,超聲-微波時間50 min,微波功率500 W,按照此參數進行驗證試驗,大青葉多糖得率為10.49%。張明以山東大青葉為材料,采用復合酶水解、乙醇沉淀法提取其中的多糖,多糖平均得率為18.24%[7],高于本研究的多糖得率10.49%,主要原因是張明在提取大青葉多糖過程中沒有進行去蛋白和透析,而且所選取的大青葉原料產地、提取實驗條件和設備差異也會造成提取率的不同。
2.1.3 超聲-微波協同提取同其他提取方法的對比
為了驗證超聲-微波協同提取效果,進行了對比提取試驗,在超聲-微波協同提取得到最佳參數下進行。對比試驗安排和結果見表2,可知僅利用超聲輔助提取多糖得率為8.11%,僅利用微波輔助提取多糖得率是8.84%,水提法多糖得率僅為7.52%,都顯著低于超聲-微波協同提取法(P<0.05)的多糖得率,說明超聲-微波協同萃取方法是有效的,可以顯著提高大青葉多糖得率。

表2 對比試驗安排和結果
注:a,b,c,d字母不同表示差異顯著(P<0.05),相同表示差異不顯著(P>0.05)。下同。
Note:The lettersa,b,c,dindicated a significant difference (P<0.05).The same below.
2.2 大青葉多糖化學成分和單糖組成
對大青葉多糖進行化學成分和單糖組成進行分析,由表3可知大青葉多糖化學組成包括63.8%總糖、13.1%蛋白質、14.2%硫酸根和12.6%糖醛酸。圖2是大青葉多糖總離子流色譜圖,同單糖標準品的出峰時間和質譜圖進行比較,由峰面積可知大青葉多糖中含量最高的是半乳糖33.1%,之后依次是阿拉伯糖24.6%、鼠李糖17.2%、葡萄糖12.0%、甘露糖6.2%、木糖4.5%、巖藻糖2.4%(表3)。
2.3 大青葉多糖的分子質量及分布
采用高效尺寸排阻色譜-多角度激光光散射儀-示差折光檢測器聯機系統對大青葉多糖的分子質量及分布情況進行了研究,示差折光檢測曲線見圖3。利用ASTRA 6.1軟件進行分析,得到分子質量(Mw)為7.85×105u,回轉半徑(Rg)為183.3 nm。
2.4 大青葉多糖的鏈接方式
利用GC-MS分析大青葉多糖鏈接方式,各碎片的質譜圖與標準質譜NIST庫和文獻[17]進行比對分析,質譜解析結果列于表4。可知殘基中主要存在2,3,4,6-四-O-、2,3,6-三-O-、2,4-二-O-甲基吡喃半乳糖醇乙酸酯,2,3,5-三-O-、3,4-二-O-、2,3-二-O-甲基阿拉伯糖醇乙酸酯,3,4-二-O-甲基吡喃鼠李糖醇乙酸酯,其峰面積分別為9.0%、14.6%、9.5%、8.8%、7.6%、7.5%、8.5%,說明大青葉多糖殘基中主要存在-(→1)-、-(1→4)-和-(1→3,6)-(鏈接的吡喃半乳糖基,-(→1)-、-(1→2)-和-(1→5)-鏈接的阿拉伯糖基,-(1→2)-鏈接的吡喃鼠李糖半乳糖基,同時也存在其它比例比較小的糖基和分支。
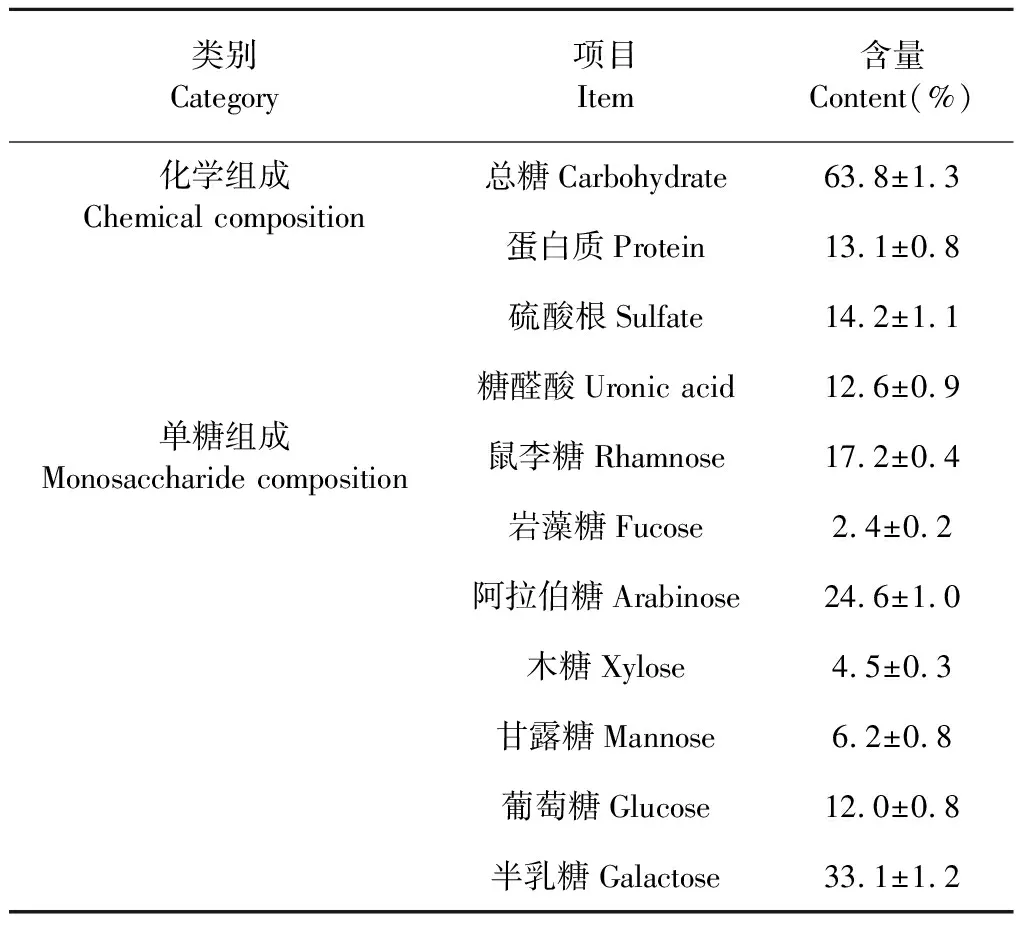
表3 大青葉多糖化學成分和單糖組成
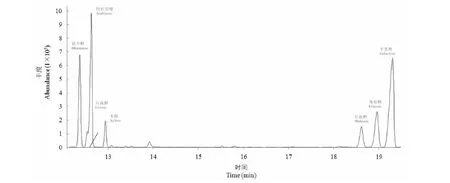
圖2 大青葉多糖糖醇乙酸酯衍生物的總離子流色譜圖Fig.2 Chromatograms of total ion current spectra for the alditol acetates derivatives of polysaccharide from Folium isatidi
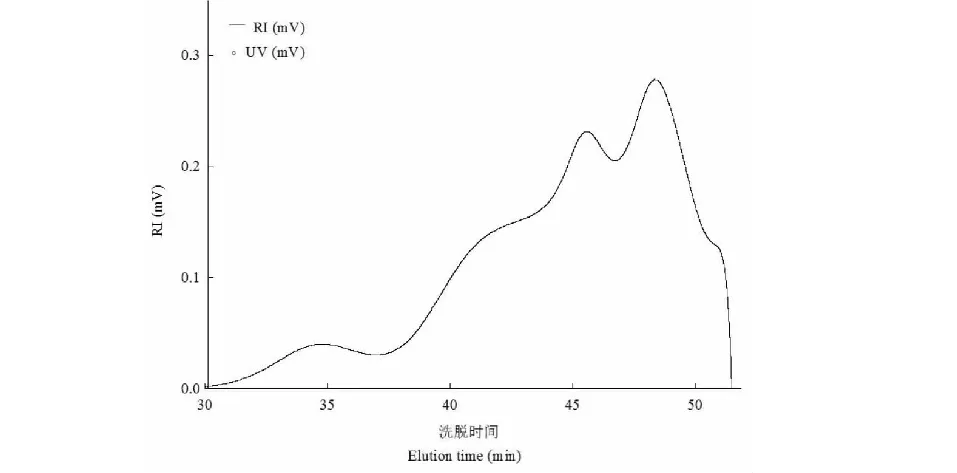
圖3 大青葉多糖示差折光檢測曲線Fig.3 RI chromatogram of polysaccharide from Folium Isatidi
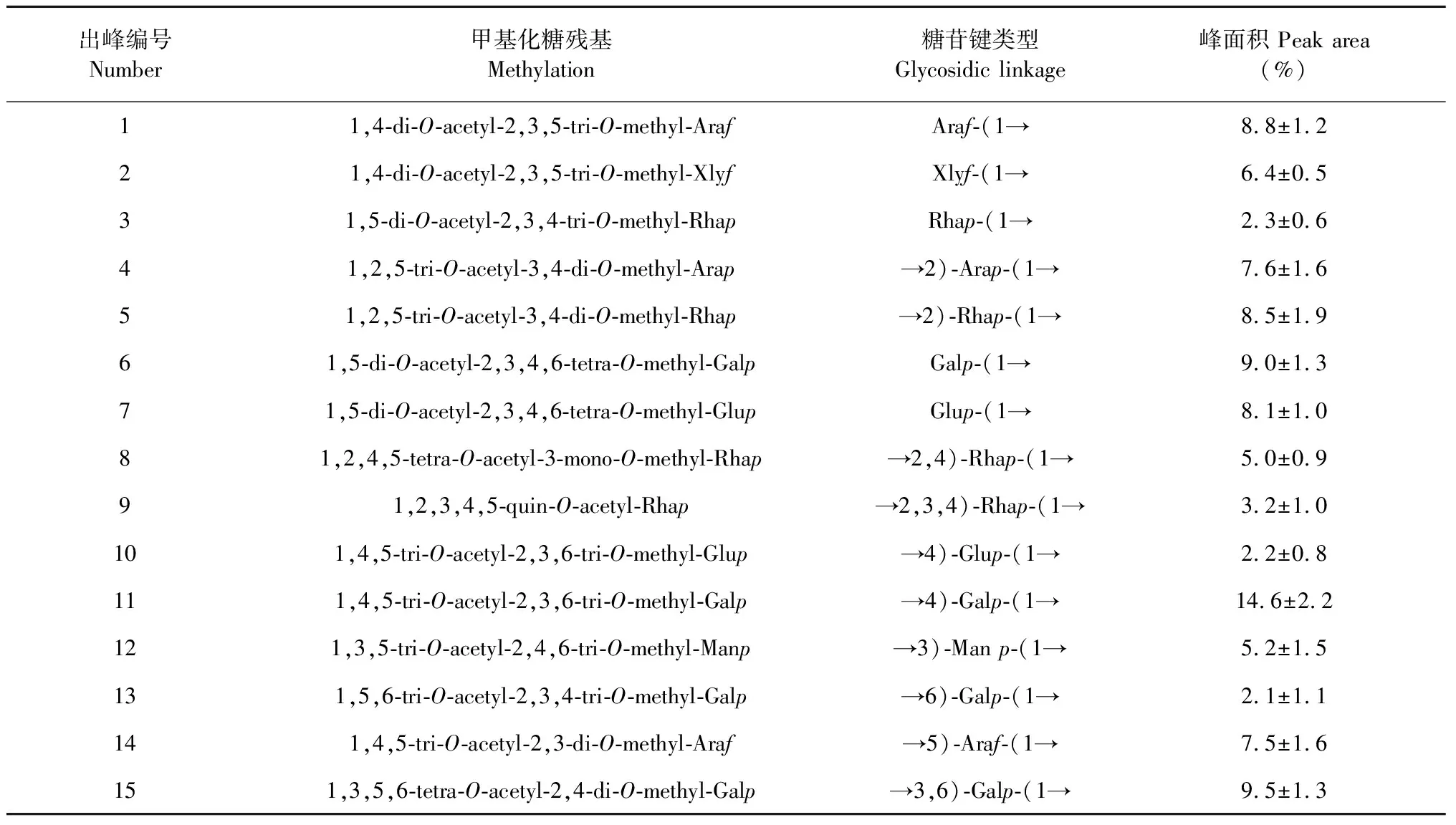
表4 大青葉多糖和還原后大青葉多糖的甲基化分析結果
2.5 大青葉多糖的紅外光譜
大青葉多糖的FT-IR圖(見圖4)給出了大青葉多糖的特征吸收峰,通過FT-IR分析可以確定為多糖化合物[18,19]。位于3 400 cm-1附近寬而強的特征峰是糖分子O-H的伸縮振動引起的,表明大青葉多糖存在分子內的氫鍵。2 920 cm-1附近的一組峰是糖分子C-H的伸縮振動引起的,1 630 cm-1附近的一組峰是由C=O的伸縮振動引起的,1 420~1 220 cm-1吸收峰是C-H的變角振動,1 100 cm-1附近的吸收峰是C-O-C環內醚中的C-O的伸縮振動和C-O-H的O-H變角振動,在1 250 cm-1附近有強吸收峰,是S=O(硫酸基)的吸收峰[20],表明大青葉多糖中含有硫酸基,這與大青葉多糖中存在14.2%硫酸根的檢測結果是吻合的。
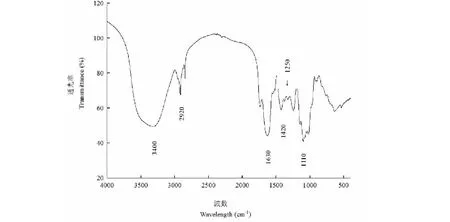
圖4 大青葉多糖的FT-IR圖Fig.4 FT-IR spectrum of polysaccharide from Folium isatidi
2.6 大青葉多糖的體外生物活性
首先分析大青葉多糖對巨噬細胞RAW264.7增殖能力的影響,大青葉多糖溶液(2、5、10 μg/mL)加入到細胞中培養后用WST-1試劑測定吸光度,同空白組相比得到巨噬細胞相對增殖率,結果見圖5。可知當大青葉多糖溶液濃度為2、5、10 μg/mL時細胞增殖率分別為100.52%、101.22%和101.85%,說明在本試驗所選定的濃度范圍內,大青葉多糖可以促進RAW264.7細胞。大青葉多糖激活RAW264.7產生NO能力結果見圖6,大青葉多糖溶液濃度為2、5、10 μg/mL時對應的NO產量分別為32.41、40.57和42.21 μmol/L,呈現劑量依賴關系,2 μg/mL的處理組與另外兩個濃度對應的NO產量差異顯著(P<0.05)。
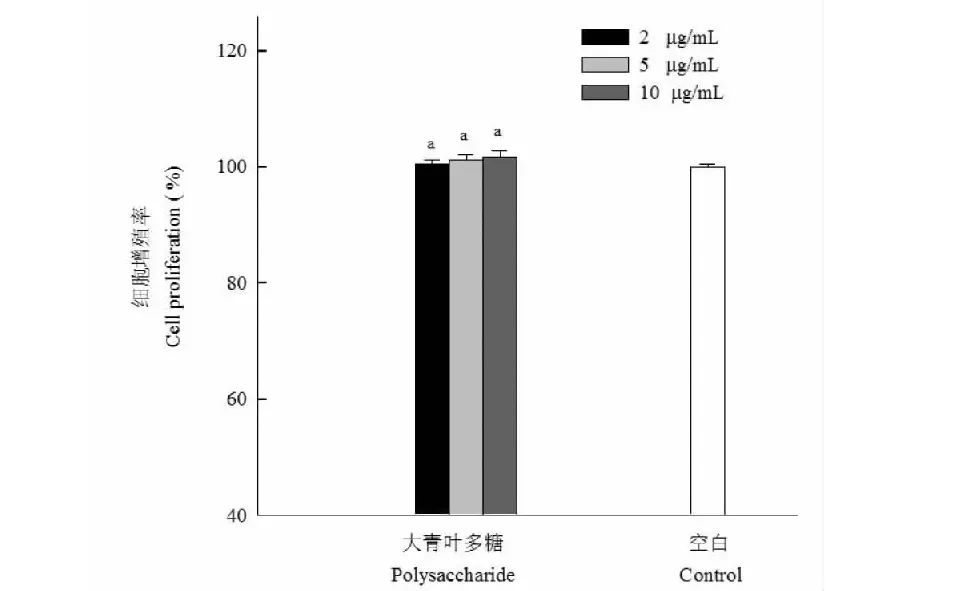
圖5 大青葉多糖對巨噬細胞增殖的影響Fig.5 Effect of polysaccharide from Folium isatidi on the proliferation of RAW264.7 cells
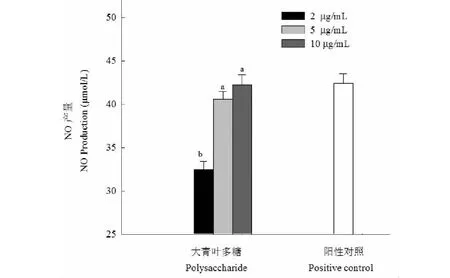
圖6 大青葉多糖對RAW246.7細胞中NO產量的影響Fig.6 The amounts of NO produced by RAW264.7 cells treatment with polysaccharide from Folium isatidi
3 結論
利用超聲-微波協同水提法可以顯著提高大青葉多的得率,提取得到的大青葉多糖中主要成分是總糖,同時還含有一定量的蛋白質、硫酸根和糖醛酸,明確了大青葉多糖的單糖組成和分子量,通過衍生化后分析了大青葉多糖的鏈接方式。體外生物活性研究表明,大青葉多糖可以促進RAW264.7細胞增殖并產生一氧化氮。本文對大青葉多糖的分子特性和生物活性的研究成果可以為大青葉多糖的研究利用提供理論基礎和參考,但對于體內外生物活性和及其機制有待于進一步研究。
