一例NPHP1純合缺失所致的Joubert綜合征
2019-07-30 07:06:48馬斯禹阮雯聰曹宗富李海峰沈玥蔡瑞琨陸超羅敏娜馬旭高華方鄒艷
生殖醫學雜志 2019年7期
關鍵詞:深度
馬斯禹,阮雯聰,曹宗富,李海峰,沈玥,蔡瑞琨,陸超,羅敏娜,馬旭,高華方,4*,鄒艷
(1.北京協和醫學院研究生院,北京 100730;2.國家衛生健康委科學技術研究所,北京 100081;3.浙江大學醫學院附屬兒童醫院,杭州 310003;4.國家人類遺傳資源中心,北京 102206;5.浙江省疾病預防控制中心,杭州 310051)
Joubert綜合征(Joubert Syndrome,MIM#213300)因Joubert等[1]于1969年首次報道而得名,是一種以小腦蚓部發育不良為主要特征的神經系統疾病,其主要的臨床表現包括陣發性呼吸急促、眼球運動障礙、肌張力減低、共濟失調等,亦可合并視網膜、腎臟、肝臟、骨骼等多器官受累[2-3],其主要的影像學特征為小腦蚓部部分或完全缺如、小腦上腳增粗和“磨牙征”(molar tooth sign,MTS)[4]。Joubert綜合征通常表現為常染色體隱性遺傳,僅Joubert綜合征10型表現為X連鎖隱性遺傳(OFD1)。該病在不同人群的發病率不一,目前缺乏中國的發病率的數據。Joubert綜合征具有明顯的遺傳異質性,超過34個基因被發現與其相關[5-9]。其中Joubert綜合征4型是因NPHP1突變導致的,患兒通常伴有腎臟損害的并發癥風險。2015年,浙江大學醫學院附屬兒童醫院康復科收治一例發育遲緩,磁共振顯示“磨牙征”、疑似Joubert綜合征的患兒,通過二代測序和MLPA等技術,明確為NPHP1純合缺失引起的Joubert綜合征,現將該患兒的臨床特點及遺傳學分析結果報道如下。
材料和方法
一、實驗材料
本研究Joubert綜合征患者來自于浙江大學醫學院附屬兒童醫院康復科。經國家衛生健康委科學技術研究所倫理委員會批準,在知情同意的前提下,收集了患兒的臨床資料,并采集患兒及其父母的靜脈血進行基因檢測。正常人的血樣(用于MLPA實驗)取自國家衛生健康委科學技術研究所門診部。
全血基因組DNA提取試劑盒購自Qiagen公司(德國);Taq酶、DNA marker 購自北京全式金生物技術有限公司;PCR擴增引物由華大六合科技有限公司合成;MLPA探針(SALSA MLPA P387NPHP1 probemix)及試劑盒(SALSA MLPA EK1-FAM reagent kit)為MRC-Holland公司(荷蘭)產品。
二、DNA提取和全外顯子組測序
按照QIAamp DNA Blood Mini Kit(Qiagen,德國)的操作說明從全血中提取基因組DNA,Qubit3.0(Invitrogen,美國)定量。取0.5 μg待測樣品DNA用Covaris 超聲儀(Covaris,美國)隨機打斷成180~280 bp的片段,用SureSelect Human All Exon V6(60 Mb) kit (Agilent,美國)進行建庫和全外顯子組捕獲,在NovaSeq 6000測序儀上(Illumina,美國)進行2×150雙端測序。測序由北京諾禾致源科技股份有限公司完成。
三、測序數據分析
下機數據經過質量控制,去除污染的、低質量的reads后得到clean data,通過以下幾個步驟進行分析:(1)以GRCh37/hg19基因組作為參考基因組,應用Burrows-Wheeler Aligner(BWA)軟件、Picard 軟件和Genome Analysis Toolkit (GATK)軟件對所有的SNP、InDel位點進行比對、分析,利用Ensembl Variant Effect Predictor對變異位點進行注釋;(2)去除內含子區突變和同義突變;(3)去除dbSNP151數據庫(http://www.ncbi.nlm.nih.gov/snp/)、gnomAD數據庫(http://gnomad.broadinstitute.org)等公共數據庫中MAF≥1%的突變;(4)優先分析已知的Joubert綜合征致病基因,篩選無義突變、移碼突變、剪接位點突變、終止密碼子丟失等對蛋白質功能影響較大的變異位點,按照ACMG的指南進行變異位點的解讀;(5)利用GATK軟件計算已知Joubert綜合征致病基因的平均測序深度,進行拷貝數變異(copy number variation,CNV)的分析;(6)利用IGV軟件查看二代測序的結果。
四、Sanger測序驗證及突變功能分析
根據全外顯子變異位點的分析結果,獲得候選致病基因AHI1,針對其編碼區所在的第4~29號外顯子區域設計引物(表1),以患兒及其父母的基因組DNA為模板進行聚合酶鏈式反應(PCR),所得產物用于ABI 3730xl(美國)的Sanger測序。使用Chromas軟件查看Sanger 測序峰圖,用SeqMan 軟件比對測序結果。
五、多重連接探針擴增(Multiplex Ligation-dependent Probe Amplification,MLPA)
根據全外顯子組平均測序深度的分析結果提示,利用多重連接探針擴增(MLPA)技術對患兒及其父母的NPHP1基因進行拷貝數變異檢測,同時用正常人的DNA進行對照實驗。具體操作步驟如下:(1)DNA變性:取100 μg DNA(5 μl)于98℃加熱5 min;(2)雜交:加入3 μl SALSA探針混合物和1.5 μl MLPA緩沖液,95℃溫浴1 min,60℃雜交16 h;(3)連接:加入32 μl連接酶混合液,54℃溫浴15 min。再于98℃加熱5 min使連接酶失活;(4)加入2 μl PCR混合物、0.5 μl聚合酶,開始PCR擴增;(5)毛細管電泳:將所得產物經86℃變性3 min以后,取0.7 μl產物混合0.2 μl LIZ500,在ABI 3730xl(美國)上進行毛細管電泳。利用Coffalyser.NET軟件對結果進行分析。
結 果
一、臨床資料
患兒,男,因“6月齡豎頭欠穩”就診。患兒系第1胎第1產,其母產前孕檢MRI顯示胎兒后顱窩池增大、小腦下蚓部發育欠佳。足月剖宮產,患兒出生時體重3 kg,身長48 cm,出生后窒息,2 h即入院,9 d后痊愈出院。
查體顯示:患兒左眼內斜,追聲能引出,追視欠靈活,逗引會笑,豎頭欠穩,不會翻身,雙手握拳,無主動抓物意識,雙下肢負重差,四肢肌力IV級,肌張力偏低。輔助檢查:肝臟、膽囊、脾臟、胰臟、雙腎、輸尿管及膀胱超聲檢查未見異常。顱腦 MRI:軸位T1W1示小腦蚓部小,中腦變形呈“磨牙”狀(圖1),第四腦室上部呈“蝙蝠翼”。診斷:Joubert綜合征。
后隨訪至今:該患兒1歲能獨坐,2歲4個月能走。現年4歲4個月,身高104 cm,體重17 kg,能簡單交流,獨立上下樓梯、穿脫衣物,手眼協調欠佳。
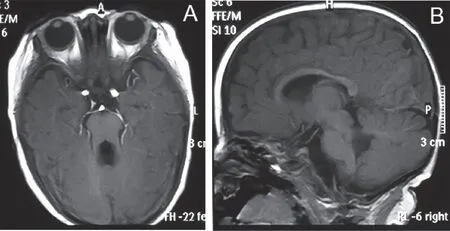
A:軸位T1W1顯示小腦蚓部發育小和“磨牙征”;B:矢狀位T1W1顯示小腦上腳抬高增粗圖1 患兒頭部MRI影像圖
二、全外顯子組測序及變異位點分析結果
全外顯子組測序產出Raw data 為19.19 G,clean data為18.75 G,總reads數為127 906 426條,Q30為95.62%。總捕獲效率為83.03%,目標捕獲區平均測序深度為160×,其中≥1×測序深度下的覆蓋度為98.23%,≥10×測序深度下的覆蓋度為97.79%,≥20×測序深度下的覆蓋度為97.41%,數據量符合分析要求。
原始數據經過比對、注釋、過濾、篩選以后,在6號染色體的AHI1基因(NM_001134831.1)上的第7號外顯子發現1個雜合的缺失突變,c.533_534delAA,p.Glu178GlyfsTer3。該位點因在533和534處連續缺失兩個堿基,造成第178位的谷氨酸改變為甘氨酸,并造成終止密碼子提前,形成截短翻譯的蛋白。該變異位點在dbSNP數據庫中的編號為rs759593846,ExAC數據庫中顯示其突變頻率為0.000 033 14,其中,東亞人中的突變頻率為0.000 463 8;GnomAD數據庫中的突變頻率為0.000 042 77,其中,東亞人中的突變頻率為0.000 614 3;ClinVar數據庫對該位點尚無記錄。根據ACMG的指南,該位點的致病性等級為可能致病,但是因為該位點為雜合突變,AHI1基因上的單一雜合位點不足以解釋患兒的致病原因。因此我們接下來采用Sanger測序法對該位點在患兒和父母的DNA中進行了驗證,并對AHI1基因的所有外顯子進行了擴增測序。
三、PCR擴增產物的Sanger測序驗證
Sanger測序結果表明,先證者AHI1基因上的移碼突變位點c.533_534delAA來自母親(圖2)。并且,在對AHI1所有外顯子進行擴增測序以后,沒有在其他外顯子及剪接位點區域發現可能的致病突變,因此考慮利用全外顯子組測序數據對已知致病基因的拷貝數變異情況進行分析。

紅色箭頭示變異位點所在的堿基圖2 AHI1基因c.533_534delAA位點的Sanger測序峰圖
四、拷貝數變異的分析
利用GATK的depth of coverage功能對患兒的全外測序數據中38個Joubert綜合征相關基因的平均測序深度進行了統計分析(圖3)。其中測序深度最高的為CELSR2,平均測序深度為262.38×,AHI1基因的平均測序深度為98.49×,而NPHP1基因的測序深度為0.24×。通過IGV軟件查看bam文件中NPHP1基因的測序情況,發現該基因區段捕獲很少測序數據,由此,我們推測患兒的NPHP1可能存在純合型缺失。
五、NPHP1拷貝數變異的驗證
采用MLPA技術對患兒及其父母的NPHP1基因進行拷貝數變異的驗證分析,同時取一個正常人的DNA進行實驗,作為陰性對照。該試劑盒的探針覆蓋了NPHP1的所有外顯子區域。結果顯示,正常人的NPHP1基因的第1~20個外顯子NPHP1為2個拷貝,不存在拷貝數變化。患兒的NPHP1基因的第1~20個外顯子全部為0拷貝,屬于NPHP1基因的純合型缺失,而患兒父親和母親的NPHP1基因的第1~20個外顯子為1個拷貝,屬于NPHP1基因的雜合型缺失(圖4)。
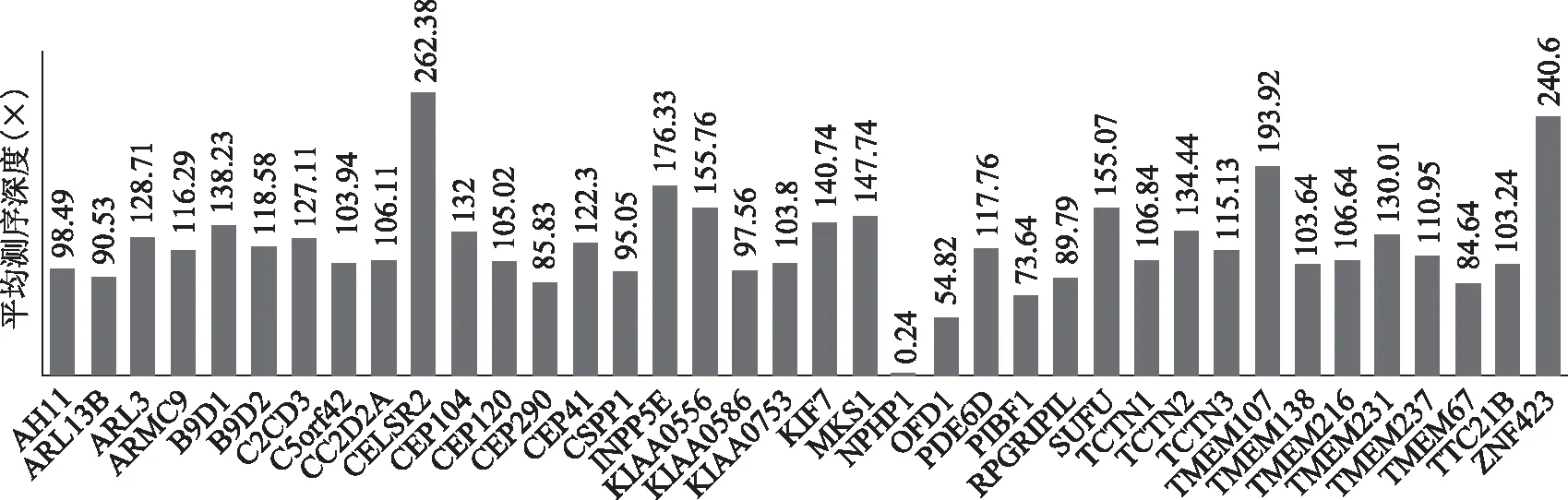
38個Joubert綜合征相關基因的平均測序深度。橫坐標為全外顯子組測序包含的38個Joubert綜合征相關基因,縱坐標為相應基因的平均測序深度(×)圖3 Joubert綜合征相關基因的平均測序深度結果圖
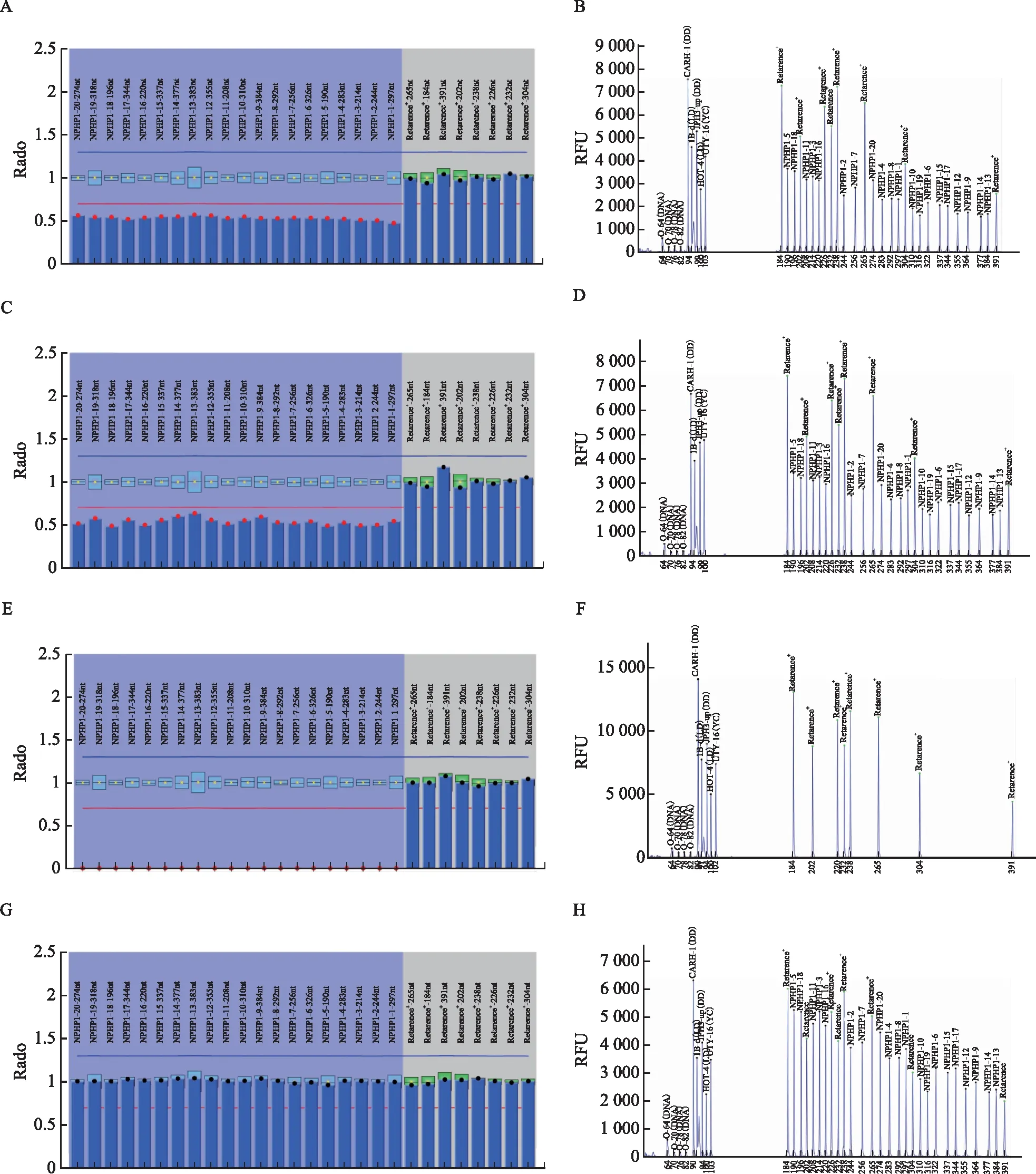
A、B(先證者父親),C、D(先證者母親):均為NPHP1基因1~20號外顯子雜合型缺失;E、F:先證者,NPHP1基因1~20號外顯子的純合型缺失;G、H:陰性對照,NPHP1基因1~20號外顯子不存在拷貝數變化圖4 MLPA分析結果
討 論
Joubert綜合征是一種以小腦發育畸形為主要特征的罕見的神經系統疾病[1]。小腦蚓部發育不全、肌張力低、發育遲緩、異常呼吸或者異常眼運動是1992年Saraiva和Baraitser制定的診斷標準[10]。1997年Maria等[11]又把磁共振成像中的“磨牙征”等加入到Joubert綜合征的診斷標準中。本研究中的患兒生后出現窒息,自幼發育落后,雙眼追視不良,四肢肌張力低,磁共振MRI顯示小腦蚓部小,且中腦呈“磨牙征”,符合Joubert綜合征的診斷標準。
Joubert綜合征通常遵循常染色體隱性遺傳的模式,僅在因OFD1基因突變導致的患兒中表現出X連鎖隱性遺傳的特點。目前已有超過34個基因被確認為Joubert綜合征的致病基因,可以解釋62%~94%受檢者的發病原因[6,12]。這些致病基因均與纖毛的功能相關,參與纖毛功能相關的信號轉導通路[13],因此,Joubert綜合征又與腎單位腎癆(Nephronophthisis,NPHP)、先天性黑矇(Leber Congenital Amaurosis,LCA)、巴德-畢德氏綜合征(Bardet-Biedl Syndrome,BBS)等其他因纖毛基因突變導致的人類疾病統稱為“纖毛相關疾病(ciliopathy)”。
我們在利用全外顯子組測序查找患兒的分子遺傳學病因的過程中,首先發現了位于AHI1基因上的一個雜合移碼突變(c.533_534delAA,p.Glu178GlyfsTer3)。但是通過對AHI1所有外顯子區域的測序分析,沒有找到其他可能的致病位點,從而開始針對Joubert綜合征的已知致病基因進行拷貝數變異的分析。經過分析,發現了NPHP1基因所在區域的測序深度均接近于0,提示該基因可能存在純合缺失。隨后,我們利用MLPA方法對患兒及其父母的NPHP1拷貝數進行了驗證,結果表明患兒確實存在NPHP1全基因的純合型缺失,父母均為NPHP1基因的雜合型缺失,符合遺傳共分離定律,最終明確了該患兒為NPHP1純合型缺失所導致的Joubert綜合征4型。
NPHP1編碼基因定位于2q13,具有20個外顯子。Hildebrandt等[14]于1997年首次發現NPHP1與青少年型腎單位腎癆(Juvenile Nephrophthisis)有關。除此之外,NPHP1基因突變可以導致Joubert綜合征4型和家族性腎視網膜營養不良1型[15](Senior-Loken Syndrome,SLS)。這三種疾病的臨床表現彼此交疊。在NPHP1的突變譜中,長達290 kb的純合缺失是其主要的突變形式,也有部分患者攜帶點突變并伴有基因的雜合型缺失[16-17]。2011年,Chaki等[18]對來自365個家系中的440個具有腎單位腎癆相關表現的纖毛疾病患者進行了基因型和表型的相關性研究,發現有來自235個家系的248名患者是由于NPHP1基因突變導致的,其中有93%(219/235)的NPHP1突變都是基因的純合缺失。這235個NPHP1突變的家系均表現出典型的青少年腎單位腎癆,終末期腎衰竭發病年齡大于4歲。其中,76.5%的家系(180/235)僅僅有腎臟受累的情況,而剩下的55個家系同時伴有其他器官的受累,涉及中樞神經系統、眼睛、肝臟和心臟。2013年,Halbritter等[19]對全世界范圍內具有腎單位腎癆相關表型的1 540名患者進行遺傳學研究,結果提示在551名具有明確分子診斷的患者中超過一半以上是因為NPHP1突變導致。盡管NPHP1是腎單位腎癆的最主要致病基因,但是在Joubert綜合征的患者中,NPHP1突變所占的比例非常小。2015年,Bachmann-Gagescu等[6]對375個Joubert綜合征家系進行了遺傳學分析,最終為279名患者找到了可能的致病病因,但其中僅有5名患者為NPHP1突變,占比1.8%。值得一提的是這5名NPHP1突變的Joubert綜合征患者都是因為NPHP1基因的純合型缺失導致的。
目前國內文獻報道具有明確基因診斷的Joubert綜合征病例并不多見,主要致病基因分別為CC2D2A[20-22]、CEP290[23-24]、OFD1[25]、TMEM67、C5orf42[26]、CSPP1[27]以及INPP5E[28-29]等,尚未有NPHP1基因突變所致Joubert綜合征的報道。對于腎單位腎癆而言,國內的研究也主要以零星的臨床病例報道為主,1982~2016年間共報道腎單位腎癆病例數120余例[30],其中由于NPHP1大片段純合缺失所導致的少年型腎單位腎癆的病例僅有10例[17,31-33],2例為男孩,發病年齡分別為9歲和12歲,均在一年內快速進展為終末期腎衰竭,另有3例存在NPHP1大片段純合缺失的患兒進入慢性腎臟病(chronic kidney diseases,CKD)V期的中位數年齡為8.5歲。由于NPHP1純合缺失與腎臟并發癥密切相關,因此在為患兒明確基因診斷以后,醫生對患兒進行了尿常規、腎功能、尿微量蛋白及B超等檢查,未發現明顯異常。考慮到本例患兒至今不足5歲,已囑咐家長密切觀察,如有多飲多尿現象及早就診,定期隨訪,爭取做到并發癥的早發現早干預。
NPHP1基因突變導致疾病的具體分子機理尚不明確,其編碼的蛋白產物是Nephrocystin-1(也稱為Nephrocystin)。該蛋白特異定位于腎單纖毛和呼吸道上皮細胞纖毛的纖毛基部(Cilia Base)至轉換區(Transition Zone,TZ),以及光感受器連接纖毛內段和外段的交界處,也定位于其他組織的細胞連接區、纖毛和細胞-基質結合位點[16,34],可調控 Par3/Par6/aPKC復合體的Par6 磷酸化狀態促進緊密連接形成[32],并參與細胞黏附、局灶性粘連復合體的形成以及細胞內信號轉導的功能。
綜上所述,本文采用全外顯子組測序技術,結合MLPA方法,證實了NPHP1基因的純合缺失是導致該Joubert綜合征患兒的致病原因。此病例的研究主要有三方面的意義:(1)盡管國內之前分別報道過CEP290、TMEM67、C5orf42、CSPP1、INPP5E以及OFD1等基因突變導致的Joubert綜合征病例,但是NPHP1基因突變所致的Joubert綜合征尚屬國內首例。(2)以往針對Joubert綜合征的臨床遺傳學研究多采用靶向基因組測序或者全外顯子組測序方法,數據分析和解讀過程中主要關注點突變,幾乎不考慮拷貝數的變化。本病例雖然也采用了全外顯子組測序,但是當AHI1基因單一雜合移碼突變不足以解釋致病原因時,考慮到了拷貝數變異的可能性,增加了對目標基因平均測序深度的分析,得到了NPHP1基因可能存在純合缺失這個重要的提示,并采用多重連接探針擴增技術(MLPA)進行驗證,最終證實了該病例是因為NPHP1整個基因的大片段純合缺失導致的,這提示我們在單基因病的數據分析過程中,要考慮多種可能的突變形式,不能僅僅局限于點突變的分析。(3)雖然此次報道的患兒目前并未表現出腎臟功能的損害,但由于其致病基因是NPHP1,該基因已被研究證明與腎臟損害直接相關,患兒被確診為Joubert綜合征4型,具有很高的腎病發病風險。臨床和遺傳學相結合所作出的癥狀前診斷不僅有助于了解患兒的預后,同時對于指導患兒的隨訪具有重要的意義。
猜你喜歡
中學生數理化·七年級數學人教版(2022年6期)2022-06-05 06:50:50
快樂學習報·教育周刊(2022年16期)2022-05-01 21:25:05
中學生數理化·七年級數學人教版(2020年11期)2020-12-14 06:59:52
藝術品鑒證.中國藝術金融(2018年8期)2019-01-14 01:14:28
藝術品鑒證.中國藝術金融(2018年10期)2019-01-08 02:44:26
藝術品鑒證.中國藝術金融(2018年6期)2019-01-08 02:43:04
藝術品鑒證.中國藝術金融(2018年12期)2018-08-26 06:03:48
新聞傳播(2016年10期)2016-09-26 12:14:59
新聞傳播(2015年10期)2015-07-18 11:05:40
交通建設與管理(2015年15期)2015-03-20 15:18:57
