白細胞介素-4負調控NF-κB通路抑制炎癥反應的機制研究
2019-11-06 02:30:06黃潔媛劉文明
天津醫藥 2019年10期
黃潔媛,劉文明
既往研究證實,炎癥反應繼發的免疫失衡是重癥感染病理生理過程的重要特點,也是重癥感染患者病情加重的重要原因[1]。肺部嚴重感染時極易誘發急性肺損傷,這與機體免疫防御機制過度激活、促炎/抑炎反應失衡和氧化應激損傷有關[2]。因此在抗感染的基礎上,恢復促炎/抑炎反應平衡,阻斷炎癥級聯反應是急性肺損傷早期治療的關鍵。白細胞介素(interleukin,IL)-4是重要的抑炎細胞因子,由CD4+T細胞分泌,對淋巴細胞的遷移具有重要作用。既往已有研究證明,IL-4可以下調炎癥反應強度,降低嚴重感染小鼠的死亡率[3],但是其抑炎機制目前尚未明了。髓樣分化因子88(myeloid differentiation factor 88,MyD88)是Toll樣受體信號通路中的重要接頭分子,可以與核因子(NF)-κB形成炎癥反應通路,導致NF-κB p65蛋白從細胞質轉入細胞核,從而誘導 IL-1、IL-6、腫瘤壞死因子(tumor necrosis factor,TNF)-α等促炎細胞因子的轉錄和表達,放大炎癥信號。MyD88/NF-κB信號通路在感染性疾病的發生和發展中占有重要地位[4-5]。已有研究證明,IL-4可以抑制IκB激酶(IKK)的活性,進而抑制NF-κB活化。另外,IL-4對NF-κB的作用不僅局限于下游水平,對上游也會造成影響[6-7]。因此筆者推測,IL-4可能作用于MyD88/NF-κB信號通路,進而影響NF-κB活性及p65蛋白的核移位以下調炎癥反應。本研究以脂多糖(lipopolysaccharides,LPS)啟動小鼠巨噬細胞株Ana-1的炎癥反應,分析IL-4預處理對LPS誘導的巨噬細胞MyD88/NF-κB信號通路的影響,探討IL-4抑制炎癥的可能機制。
1 材料與方法
1.1主要材料和儀器 小鼠巨噬細胞Ana-1購自上海信裕生物科技有限公司(貨號XY-M001);小鼠IL-4購自上海恒斐生物科技有限公司(貨號130-097-760);DMEM培養基購自杭州仟諾生物科技有限公司;胎牛血清購自杭州四季青生物工程有限公司;兔抗鼠MyD88和NF-κB p65單克隆抗體購自上海生物工程有限公司;羊抗兔生物素化IgG購自北京中杉金橋公司;小鼠TNF-α和IL-6酶聯免疫吸附法(ELISA)檢測試劑盒購自上海哈靈生物科技公司;小鼠NF-κB p65 ELISA檢測試劑盒購自上海酶聯生物科技公司;胞漿和胞核蛋白提取試劑盒購于武漢博士德有限公司。ABI 7500實時熒光定量PCR儀(ABI),半干轉印儀(Bio-Rad),垂直電泳儀(Bio-Rad),HBS-1096A酶標儀(南京德鐵實驗設備有限公司)。
1.2方法
1.2.1細胞培養和構建LPS±IL-4處理Ana-1細胞模型 Ana-1巨噬細胞采用DMEM培養基培養,內含10%滅活胎牛血清、100 U/mL鏈霉素和100 mg/L青霉素,37℃,5%CO2常規細胞培養,隔日換液。當細胞貼壁融合度達到90%以上時,用0.25%胰蛋白酶進行傳代。傳代細胞接種于6孔板(1×106個/孔)。取對數生長期的Ana-1巨噬細胞,將6孔板中的Ana-1細胞分為2組:分別為LPS組(給予50μg/L LPS刺激)、LPS+IL-4組(10μg/L IL-4預培養1 h后給予LPS刺激)。在0、0.5、1和2 h時收集細胞培養上清液,用于后續實驗。
1.2.2RT-PCR檢測MyD88、NF-κB mRNA表達水平 TRIzol法提取Ana-1巨噬細胞總RNA,按逆轉錄試劑盒說明將其逆轉錄為cDNA。以β-actin為內參,通過ABI 7500實時熒光定量PCR儀分別進行熒光定量PCR反應。引物由上海生工生物工程股份有限公司合成并提供,序列見表1。RT-PCR反應條件為:預變性95℃30 s;95℃5 s,60℃30 s,共計40個循環。每個樣本至少重復3次。2-ΔΔCt法計算mRNA的相對表達量。
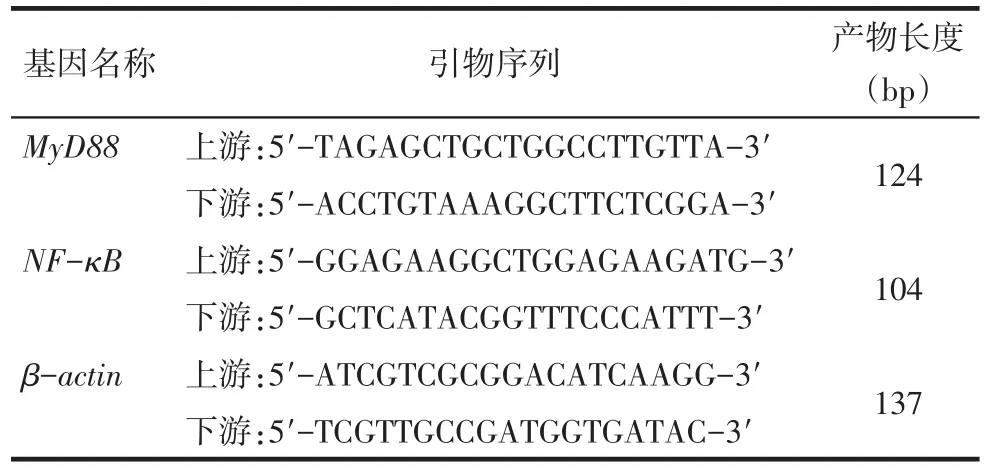
Tab.1 The primer sequence of MyD88,NF-κB and actin表1 RT-PCR引物序列
1.2.3Western blot檢測MyD88和NF-κB蛋白表達水平 按照試劑盒說明書分別提取總蛋白、胞漿蛋白和胞核蛋白,BCA法進行蛋白定量。取20μL樣品加入5×SDS緩沖液,煮沸5 min使蛋白變性。經十二烷基磺酸鈉-聚丙烯酰胺凝膠電泳(SDS-PAGE)后將蛋白轉移至PVDF膜上,5%脫脂奶粉封閉1 h,孵育一抗(兔抗鼠MyD88或NF-κB p65單克隆抗體,1∶1 000),4 ℃過夜,棄去一抗,TBST洗膜3次,每次5 min。室溫孵育二抗(羊抗兔生物素化IgG,1∶1 000)2 h,棄去二抗,TBST洗膜3次,每次5 min。ECL顯色,同期以β-actin作內參。顯影后通過Quantity One軟件分析各條帶,以各條帶與內參吸光度的比值表示MyD88/NF-κB p65的相對表達量。
1.2.4ELISA檢測NF-κB p65胞核/胞漿比例 按照試劑盒說明書分別提取胞漿蛋白和胞核蛋白,BCA法進行蛋白定量。按照NF-κB p65 ELISA檢測試劑盒說明書分別檢測NF-κB p65在細胞核及細胞漿中的含量,檢測時波長設置為450 nm,并計算NF-κB p65胞核/胞漿比例。
1.2.5ELISA檢測IL-6和TNF-α含量 收集各組細胞的培養液,3 000 r/min離心10 min,取上清液,按照TNF-α和IL-6 ELISA檢測試劑盒說明書進行IL-6和TNF-α含量的檢測,檢測波長設置為450 nm。
1.3統計學方法 采用SPSS 17.0統計學軟件進行數據分析。符合正態分布的計量數據以均數±標準差(±s)表示,2組不同時間點間比較采用重復測量設計的方差分析,同一時間點組間比較行t檢驗。P<0.05為差異有統計學意義。
2 結果
2.1IL-4對LPS活化的Ana-1細胞表達MyD88的影響 隨著Ana-1細胞培養時間的延長,LPS組MyD88 mRNA和蛋白表達水平均呈逐漸升高趨勢(P<0.05)。而LPS+IL-4組MyD88 mRNA表達水平無明顯變化(P>0.05),MyD88蛋白表達水平則呈先升高后下降趨勢(在0.5 h時最高,2 h時下降至最低)。LPS+IL-4組MyD88 mRNA和蛋白表達水平在0 h和0.5 h時與LPS組差異無統計學意義(P>0.05),在1 h和2 h時顯著低于LPS組(P<0.05)。見圖1、表2。
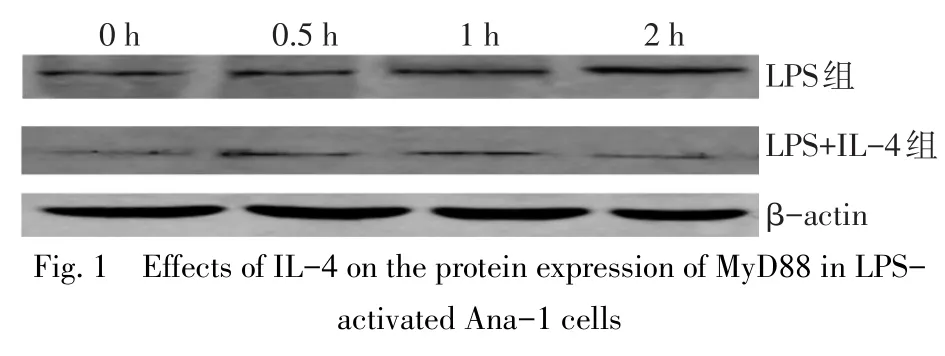
圖1 IL-4對LPS活化的Ana-1細胞表達MyD88蛋白的影響
2.2IL-4對LPS活化的Ana-1細胞NF-κB表達的影響 隨著Ana-1細胞培養時間的延長,無論是LPS組還是LPS+IL-4組,NF-κB mRNA和蛋白表達水平均呈現明顯升高趨勢(P<0.05)。而2組不同時間點NF-κB mRNA和蛋白表達水平比較差異均無統計學意義(P>0.05)。見圖2、表3。
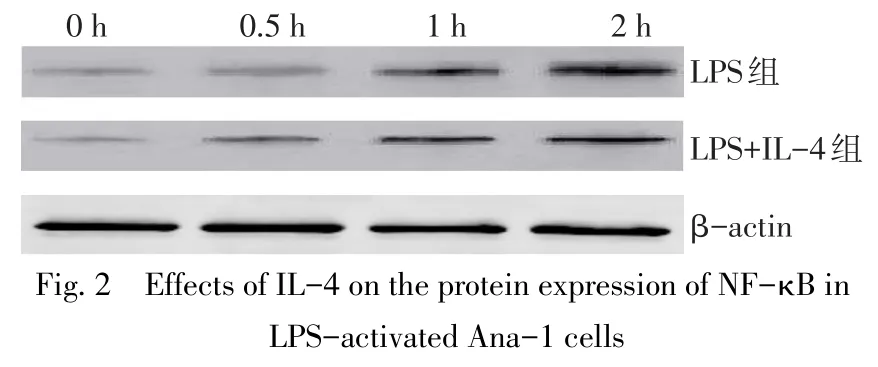
圖2 IL-4對LPS活化的Ana-1細胞表達NF-κB蛋白的影響
Tab.2 Effects of IL-4 on the mRNA and protein expression of MyD88 in LPS-activated Ana-1 cells表2 IL-4對LPS活化的Ana-1細胞表達MyD88 mRNA和蛋白的影響 (n=5,±s)

Tab.2 Effects of IL-4 on the mRNA and protein expression of MyD88 in LPS-activated Ana-1 cells表2 IL-4對LPS活化的Ana-1細胞表達MyD88 mRNA和蛋白的影響 (n=5,±s)
*P<0.05;a與0 h比較,b與0.5 h比較,c與1 h比較,P<0.05
組別LPS組LPS+IL-4組t MyD88 mRNA 0 h 1.30±0.34 1.26±0.48 0.152 0.5 h 1.68±0.12 1.72±0.43 0.200 1 h 2.14±0.30a 1.52±0.21 3.786*2 h 2.67±0.43ab 1.21±0.14 3.063*F 10.372*1.415 MyD88蛋白(%)0 h 7.36±0.61 7.29±1.01 0.133 0.5 h 15.37±0.44a 16.01±0.84a 1.509 1 h 17.00±0.81ab 12.64±0.32ab 11.942*2 h 19.37±1.24abc 7.01±0.97abc 17.555*F 117.998*82.627*
Tab.3 Effects of IL-4 on the mRNA and protein expression of NF-κB in LPS-activated Ana-1 cells表3 IL-4對LPS活化的Ana-1細胞表達NF-κB mRNA和蛋白的影響 (n=5,±s)
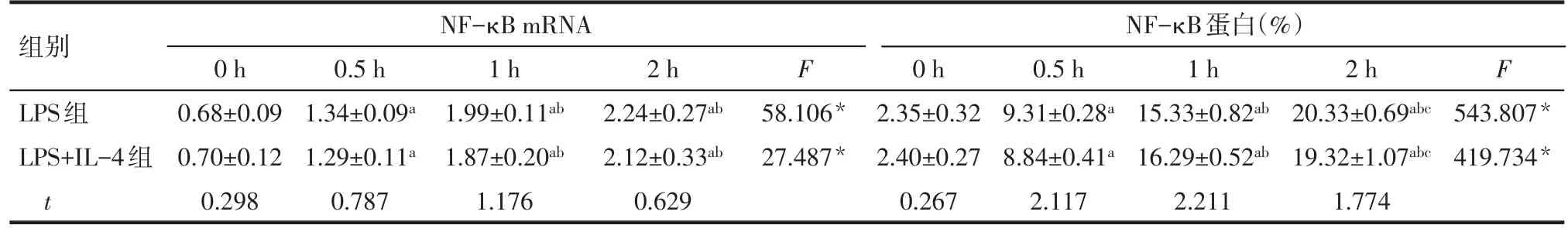
Tab.3 Effects of IL-4 on the mRNA and protein expression of NF-κB in LPS-activated Ana-1 cells表3 IL-4對LPS活化的Ana-1細胞表達NF-κB mRNA和蛋白的影響 (n=5,±s)
*P<0.05;a與0 h比較,b與0.5 h比較,c與1 h比較,P<0.05
組別LPS組LPS+IL-4組t NF-κB mRNA 0 h 0.68±0.09 0.70±0.12 0.298 0.5 h 1.34±0.09a 1.29±0.11a 0.787 1 h 1.99±0.11ab 1.87±0.20ab 1.176 2 h 2.24±0.27ab 2.12±0.33ab 0.629 F 58.106*27.487*NF-κB蛋白(%)0 h 2.35±0.32 2.40±0.27 0.267 0.5 h 9.31±0.28a 8.84±0.41a 2.117 1 h 15.33±0.82ab 16.29±0.52ab 2.211 2 h 20.33±0.69abc 19.32±1.07abc 1.774 F 543.807*419.734*
2.3NF-κB p65胞核/胞漿比例變化 隨著Ana-1細胞培養時間的延長,LPS組NF-κB p65胞核/胞漿比例呈逐漸上升的趨勢(P<0.05);而LPS+IL-4組呈先上升后下降的趨勢(P<0.05)。LPS+IL-4組NF-κB p65胞核/胞漿比例在0 h和0.5 h時與LPS組差異無統計學意義(P>0.05),在1 h和2 h時明顯低于LPS組(P<0.05),見表4。
2.4IL-4對LPS誘導的IL-6和TNF-α水平的影響 隨著Ana-1細胞培養時間的延長,LPS組IL-6和TNF-α水平呈逐漸升高的趨勢(P<0.05);LPS+IL-4組則呈先升高后下降的趨勢(P<0.05)。LPS+IL-4組IL-6和TNF-α水平在0 h和0.5 h時與LPS組差異無統計學意義(P>0.05),在1 h和2 h時明顯低于LPS組(P<0.05),見表5。
Tab.4 Changes of nuclear/cytoplasmic ratio of NF-κB p65表4 NF-κB p65胞核/胞漿比例變化 (n=5,±s)
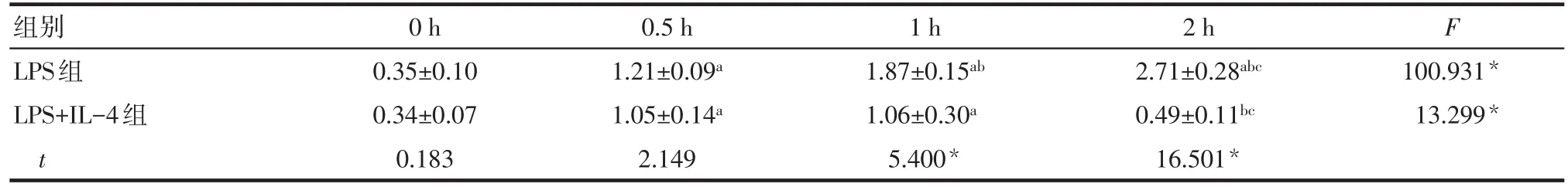
Tab.4 Changes of nuclear/cytoplasmic ratio of NF-κB p65表4 NF-κB p65胞核/胞漿比例變化 (n=5,±s)
*P<0.05;a與0 h比較,b與0.5 h比較,c與1 h比較,P<0.05
組別LPS組LPS+IL-4組t 0 h 0.35±0.10 0.34±0.07 0.183 0.5 h 1.21±0.09a 1.05±0.14a 2.149 1 h 1.87±0.15ab 1.06±0.30a 5.400*2 h 2.71±0.28abc 0.49±0.11bc 16.501*F 100.931*13.299*
Tab.5 Effects of IL-4 on the levels of IL-6 and TNF-α in LPS-activated Ana-1 cells表5 IL-4對LPS誘導的Ana-1細胞中IL-6和TNF-α蛋白表達水平的影響 (ng/L,n=5,±s)

Tab.5 Effects of IL-4 on the levels of IL-6 and TNF-α in LPS-activated Ana-1 cells表5 IL-4對LPS誘導的Ana-1細胞中IL-6和TNF-α蛋白表達水平的影響 (ng/L,n=5,±s)
*P<0.05;a與0 h比較,b與0.5 h比較,c與1 h比較,P<0.05
組別LPS組LPS+IL-4組t IL-6 0 h 60.21±22.58 60.08±18.37 0.01 0.5 h 193.26±17.68a 189.47±24.31a 0.282 1 h 245.36±22.58ab 164.21±15.24a 6.665*2 h 276.34±26.00ab 142.36±18.67ab 9.359*F 56.847*24.972*TNF-α 0 h 47.25±8.31 46.85±6.37 0.085 0.5 h 211.64±24.80a 208.36±30.33a 0.187 1 h 298.31±40.02ab 176.34±18.64a 6.155*2 h 381.11±21.22abc 163.24±14.08ab 19.13*F 89.495*39.592*
3 討論
抗炎細胞因子IL-4主要由CD4+T細胞產生的,可以促進T淋巴細胞活性并增強巨噬細胞吞噬功能。研究表明,IL-4對B細胞、T細胞、肥大細胞和單核巨噬細胞都有免疫調節作用[8]。有研究發現,重癥肺炎患者血清中IL-4水平明顯升高,提示IL-4可能參與了機體的抗炎過程[9-10],但其具體抗炎機制尚未明確。
本研究采用LPS刺激Ana-1細胞以激活NF-κB炎癥信號通路,結果顯示,在LPS的誘導下,小鼠炎癥反應細胞模型建立成功:MyD88和NF-κB的mRNA和蛋白表達水平均明顯升高,NF-κB p65蛋白核移位增加,同時上調了細胞中IL-6和TNF-α的表達,這與以往文獻報道的LPS誘導發生的炎癥反應表現一致[11-12]。Toll樣受體4(TLR4)/NF-κB信號通路是LPS胞內信號轉導的關鍵途徑。LPS通過與TLR4結合,從而誘導TLR4聚合使得信號轉導至胞內,胞內TIR區與MyD88的羧基端結合,同時MyD88通過氨基端的死亡域與白細胞介素1受體相關激酶(IRAK)的死亡結構域結合,激活IRAK的自身磷酸化,獲得游離的IRAK繼而激活腫瘤壞死因子受體相關因子-6(TRAF-6),TRAF-6激活NF-κB二聚體的抑制蛋白家族(IκB)激酶(IKKs)。IκB由于磷酸化、泛素化后降解,使得NF-κB從靜息狀態下受IκB結合處于的抑制狀態解除,進而導致NF-κB p65轉入細胞核中誘導基因轉錄,啟動IL-6、TNF-α等促炎細胞因子基因的表達[13-15]。
Lai等[16]研究顯示,IL-4可通過抑制炎癥細胞因子的基因轉錄,影響其mRNA的穩定性,進而下調炎癥細胞因子的表達。本研究通過加入IL-4進行干預LPS誘導的炎癥反應細胞模型,結果顯示,在IL-4干預下,MyD88的mRNA和蛋白表達水平呈先升高后下降的趨勢,提示LPS誘導下NF-κB激活導致p65入核增加,使得NF-κB p65胞核/胞漿比例增高;在IL-4的干擾下,進入胞核的p65蛋白減小,活化受到抑制,從而導致NF-κB p65胞核/胞漿比例的下調。
此外,本研究結果顯示,在IL-4的干擾下,促炎細胞因子IL-6和TNF-α表達水平也隨之出現逆轉。而IL-4作用下的TLR4/NF-κB信號通路中只有MyD88發生顯著變化,提示IL-4可能通過抑制MyD88而下調NF-κB活性,從而影響IL-6和TNF-α的表達,抑制LPS啟動的炎癥反應。MyD88作為TLR4/NF-κB信號通路中的關鍵接頭分子,已有研究表明在MyD88基因敲除小鼠中,LPS并不能使血清中IL-6和TNF-α水平增加[17],提示MyD88在LPS誘導產生炎癥反應中發揮著重要作用,而本研究結果進一步提示IL-4可能作用于MyD88而參與抗炎反應。
綜上,本研究通過構建IL-4干擾的LPS誘導炎癥反應細胞模型,發現IL-4可以逆轉由于LPS誘導的促炎細胞因子IL-6和TNF-α的表達上調,可能具有抗炎作用。此外通過檢測MyD88/NF-κB信號通路中MyD88和NF-κB的表達和NF-κB p65的核移位,發現IL-4參與的抗炎機制可能與該通路中MyD88的表達下調及NF-κB p65蛋白的核移位有關。但其具體機制還需要進一步研究。
