Th17細胞在類風濕關節炎發病機制中的作用研究進展
2020-03-20 00:25:08李鑫,蔡雄,劉良
中國藥理學通報 2020年3期
李 鑫,蔡 雄,劉 良
(湖南中醫藥大學1.中醫診斷學湖南省重點實驗室、2.中藥粉體與創新藥物省部共建國家重點實驗室培育基地,湖南 長沙 410208;3. 澳門科技大學中藥質量研究國家重點實驗室,澳門 氹仔 999078)
類風濕關節炎(rheumatoid arthritis,RA)是一種慢性炎癥性自身免疫疾病,其主要病理特征為進行性關節滑膜炎癥,軟骨和骨破壞,最終造成關節畸形和功能喪失[1]。研究表明,RA是以大量CD4+T細胞浸潤為主的慢性滑膜炎癥反應,CD4+T細胞作為效應T細胞的重要成分,參與免疫應答過程的各個階段,其介導的免疫反應異常被認為是RA主要發病機制之一[2]。根據其產生的細胞因子及其功能,通常將CD4+T 細胞分為輔助性T細胞1 (helper T 1,Th1)、Th2、Th17和調節性T細胞 (regulatory T cell,Treg)四大亞群。Th17細胞主要分泌IL-17、IL-21等炎性細胞因子,具有促進炎性反應和促進關節軟骨和骨破壞進程等多重效應。因此,本文擬收集近年來Th 17細胞參與RA發病的相關研究進展并進行綜述,以期為RA病理機制研究提供參考。
1 Th17細胞概述
在免疫應答的不同階段,因抗原性質、局部微環境等因素的綜合效應,CD4+T細胞會朝不同方向分化,進而發揮其特定的生物學效應。Th17 細胞是CD4+T細胞新亞群,其主要分泌白介素IL-17A、IL-17F、IL-21等細胞因子,并表達轉錄因子維甲酸相關孤核受體(retinoid acid-related orphan receptorγ,RORγt)和RORα,而在自身免疫疾病中發揮重要作用。
在樹突細胞、單核細胞等抗原呈遞細胞作用下,IL-6協同低濃度的轉化生長因子-β(transforming growth factor β,TGF-β),激活JAK1,進而招募信號傳導及轉錄激活因子-3 (signal transducer and activator of transcription-3,STAT3),并使其磷酸化,活化的JAK1/STAT3通路激活RORγt,并促進IL-21產生[3]。在IL-6和IL-21協同作用下,產生IL-23R、RORα 和RORγt,使Naive CD4+T細胞向Th17細胞分化[4],并抑制轉錄因子叉頭蛋白3 (forkhead box protein 3,Foxp3)的負向調節作用。Th17細胞被轉錄因子STAT4和STAT6調控,生成 IL-17,同時可產生IL-23、IL-6 和腫瘤壞死因子-α (tumor necrosis factorα,TNF-α)等炎性細胞因子,IL-23可促進激活的記憶細胞產生IL-17,使Th17細胞得以存活和維持功能[5]。見Fig 1。
2 Th17細胞在RA滑膜炎癥反應中的作用
RA早期主要表現為大量CD4+T細胞浸潤為主的慢性滑膜炎癥反應。IL-17是RA早期重要細胞之一,具有強大的促炎效應,大量、廣泛表達于在RA患者關節滑液、滑膜組織、外周血、外周淋巴結等處[6]。RA患者發病前外周血IL-17表達水平顯著高于發病后,RA患者(病程<12月)外周血IL-17表達量相當高,且與C-反應蛋白(C-reactive protein,CRP)呈正相關[7]。RA患者血清Th17水平顯著升高,且與疾病活動性評分28 (disease activity score 28,DAS28)呈正相關[8]。RA患者外周血Th 17、IL-6、IL-17、IL-21表達量隨疾病活動程度增加而增加[9]。RA患者外周血單個核細胞Th17細胞中趨化因子受體6(chemokine receptors 6,CCR6)和RORγt表達量顯著增加,且關節滑液單核細胞Th17細胞比例顯著高于外周血單個核細胞[10]。樹突細胞可分泌IL-In the presence of antigen-presenting cells such as dendritic cells and monocytes,IL-6 is synergized with low-concentration TGF-β to activate JAK1,and then activated JAK1 recruits and phosphorates STAT3. JAK1/STAT3 signaling activates RORγt and promotes production of IL-21.Under the circumstance of abundant IL-6 and IL-21,naive CD4+T cells differentiate into Th17 cells,and simultaneously negative regulation of Foxp3 to this differentiation is inhibited. Transcription factors STAT4 and STAT6 regulate Th17 cells to produce pro-inflammatory cytokines IL-23,IL-6,and TNF-α.IL-23 promotes activated memory CD4+T cells to produce IL-17 which in turn maintain the survival and function of Th17 cells.
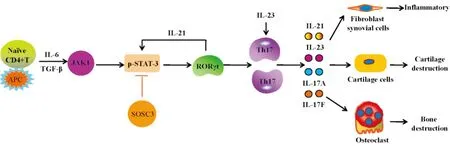
Fig 1 Pathway of differentiation of naive CD4+ T cells into Th17 cells
23促進IL-17的增殖,而參與RA風濕結節的形成[5]。膠原性關節炎(collagen induced arthritis,CIA)大鼠關節滑膜及外周血IL-17顯著升高,外周血RORγt顯著增加[11]。CIA小鼠血漿IL-17、外周血和脾臟Th17細胞比例,CD4+T細胞IL-17 mRNA表達水平均顯著增加[12]。IL-17可協同IL-1β和TNF-α共同誘導T細胞和樹突細胞的趨化[13]。IL-17協同TNF-α促進成纖維樣滑膜細胞(fibroblast-like synoviocytes,FLSs)和上皮細胞大量分泌IL-6、IL-8、前列腺素E2(prostaglandin E,PGE2)和中性粒細胞化學引誘物[13]。上述研究表明,Th17細胞在外周血、關節滑液等部位均有表達,不僅參與了RA發病,而且與疾病活動程度及臨床檢測指標密切相關。
IL-21是Th17細胞分泌的促炎性細胞因子,大量表達于CD4+T細胞及FLSs。RA患者關節滑液中CD4+IL-21+T細胞比例顯著增加,外周血CD4+IL-21+T細胞及血清IL-21與DAS28、抗-CCP抗體、血沉和類風濕因子呈正相關[14,15]。IL-21通過抑制Foxp3表達,上調RORC而促進Th17細胞分化與增殖[16]。研究顯示,聯合阻斷IL-6和IL-21可有效抑制CIA小鼠脾臟CD4+Th17細胞分化,進而減輕疾病嚴重程度[4]。IL-21/IL-21R可介導磷脂酰肌醇3-激酶/蛋白激酶B (phosphoinosmde-3-kinase/protein kinase B,PI3K/PKB) PI3K/Akt促進IL-23表達而促進RA-FLS異常增殖[17]。IL-21通過絲裂原活化蛋白激酶 (mitogen-activated protein kinase,MAPK)、STAT3及PI3K/Akt通路介導FLS分泌TNF-α、IL-6等促炎性細胞因子[18]。
3 Th17細胞在RA骨破壞中的作用
關節軟骨的破壞及骨侵蝕是RA進展期重要組織學特征。研究表明,破骨細胞是介導RA骨破壞的關鍵細胞,核因子-κB受體活化因子配基 (receptor activator of nuclear factor-κ B ligand,RANKL)是破骨細胞生成的啟動信號。CD4+T細胞介導的免疫應答在RA骨破壞中具有重要作用。IL-17A/F通過上調Runt相關轉錄因子-2 (runt-related transcription factor,RUNX2)蛋白,增強IL-6蛋白和基質金屬蛋白酶-3 (matrix metalloproteinase-3,MMP-3)mRNA表達而抑制RA關節軟骨的產生潛能[19]。骨橋蛋白可促進成骨細胞中IL-17表達而引起CIA小鼠骨質侵蝕[20]。IL-17通過活化IL-17/IL-17R/STATS-3信號通路促進RANKL表達,抑制骨保護素 (osteoprotegerin,OPG)表達而介導佐劑性關節炎(adjuvant induced arthritis,AIA)大鼠FLS破骨細胞分化[21]。RA患者骨髓血漿中IL-17A/F及CD3+CD4+IL-17+細胞百分比較骨關節炎患者顯著升高,提示骨髓微環境可以促進Th17細胞和IL-17A/F過度產生而介導RA破壞[4]。IL-17 促進K/BxN小鼠體內人分泌型卷曲相關蛋白 1 (secreted frizzled related protein,sFRP1) 分泌,減少 sFRP3 的分泌,進而抑Wnt/β-catenin/RUNX2 通路,負調控成骨細胞生成[22]。IL-17通過誘導軟骨MMP-3表達而抑制關節滑膜和軟骨細胞COL2A1表達而促進骨破壞[23]。綜上,IL-17主要通過以下途徑參與RA骨破壞:(1)促進RANKL表達,進而促進破骨細胞分化;(2)促進MMPs表達,進而降解骨細胞外基質,引起骨破壞;(3)抑制成骨細胞生成。見Fig 2。
IL-21促進FLS的RANKL表達而介導破骨細胞生成[24]。IL-21促進FLS分泌介導RA骨與關節軟骨破壞的關鍵蛋白酶MMP1、MMP2、MMP3、MMP9、MMP13而介導,其中MMP3、MMP9的活化與PI3K、 STAT-3及細胞外調節蛋白激酶 (extracellular regulated protein kinase 1/2,ERK1/2)有關[25]。IL-21/IL-21R相互作用可活化PI3K/Akt通路介導活化T細胞核因子1蛋白 (nuclear factor of activated T-cells,NFATc1)及骨吸收相關因子組織蛋白酶k (cathepsin K)和抗酒石酸酸性磷酸酶 (tartrate-resistant acid phosphatase,TRAP)表達進而介導骨破壞[17]。綜上,IL-21參與RA骨破壞的途徑主要有:(1)促進MMPs表達,進而降解骨細胞外基質,引起骨破壞;(2)調節骨吸收相關因子的表達;(3)促進破骨細胞分化。見Fig 2。

Fig 2 Bone destruction in RA promoted by Th17 cells
Th17 cells produce IL-17 and IL-21 which play an important role in bone destruction of patients with RA. IL-17 promotes bone destruction in RA through the following pathways: (1) IL-17 promotes RANKL expression and osteoclast differentiation; (2) IL-17 promotes the expression of MMPs that degrade the extracellular matrix of bone; (3) IL-17 inhibits osteoblastogenesis.IL-21 promotes bone destruction in RA through the following pathways:(1) IL-21 promotes the expression of MMPs which degrade the extracellular matrix of bone; (2) IL-21 promotes the expression of bone resorption-related factors; (3) IL-21 promotes osteoclast differentiation
4 總結與展望
綜上所述,Th17細胞及其分泌的IL-17、IL-21等促炎性細胞因子對RA早期的滑膜炎癥反應和RA晚期的骨破壞等病理環節均發揮促進作用,但對于不同病理環節的作用強度是否有差異;對于不同環節的作用機制及其效應靶點是否異同,均尚有待于進一步深入研究。因此,深入研究Th17細胞對RA不同病理環節的影響,有助于進一步揭示其針對RA不同病理環節的病理機制及其效應靶點,進而開發基于Th17細胞的RA多靶向防治藥物,具有重大的研究意義和廣袤的應用開發前景。
