揮發性有機化合物催化氧化技術
2020-05-29 12:07:40張勝碩武燕娟雍曉靜張安貴
工業催化 2020年4期
張勝碩,武燕娟,張 偉,雍曉靜,張安貴
(國家能源集團寧夏煤業有限責任公司煤炭化學工業技術研究院,寧夏 銀川 750411)
揮發性有機化合物(VOCs)是指在20 ℃、一個大氣壓(0.1 MPa)下,飽和蒸汽壓超過10.3 Pa,并且參與大氣光化學反應的有機化合物,主要是煤化工、石油化工、精細化工以及制藥、涂料、印刷、垃圾除臭等行業產生,包括非甲烷烴類、含氧有機物、含氮有機物和含硫有機物等,在室溫下很容易揮發,是形成臭氧(O3)和細顆粒污染物(PM2.5)的重要前驅體[1-3]。
《“十三五”揮發性有機物污染防治工作方案》指出,以改善環境質量為核心,以重點地區和重點行業為主要控制對象,以削減活性強VOCs組分為著力點,以全面實施固定污染源排污許可管理為抓手,以強化基礎能力建設和加強政策支持為保障,通過采取源頭削減、過程控制、末端治理全過程防控措施,全面加強VOCs污染防治,促進環境空氣質量持續改善和產業綠色轉型發展。在企業發展的同時,加強環保管理,堅決打贏藍天保衛戰。所以,全面有效治理VOCs排放勢在必行。
催化氧化技術因其具有起燃溫度低、處理效率高、適用范圍廣以及無二次污染等特點,是去除VOCs工藝中的關鍵步驟[4]。
本文對揮發性有機化合物催化氧化技術進行綜述。
1 反應機理
催化氧化VOCs已經被廣泛研究,Hermia J等[5]建立了VOCs可氧化性和分子量之間的相關性,研究表明,分子量越大,VOCs更加不容易被氧化。這個相關性與Tichenor B A等[6]提出的VOCs氧化難易程度相同,即醇<醛<芳烴<酮<乙酸酯<烷烴。
反應過程如式(1)所示,在一定反應溫度下,利用催化劑使有機氣體被催化氧化,發生無火焰燃燒,主要得到CO2和H2O,同時放出大量的熱能[7]。
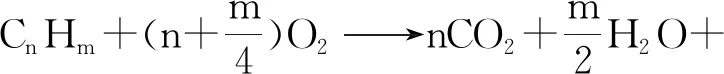
(1)
研究表明,揮發性有機化合物完全催化氧化的機理通常分為:Mars-van Krevelen(MVK)模型、Langmuir-Hinshelwood(L-H)模型和Eley-Rideal(E-R)模型。
MVK模型[8]認為,催化氧化反應發生在吸附的VOCs和催化劑晶格氧之間,而不是與氣相中的氧氣發生反應,MVK模型機理如圖1所示[9]。
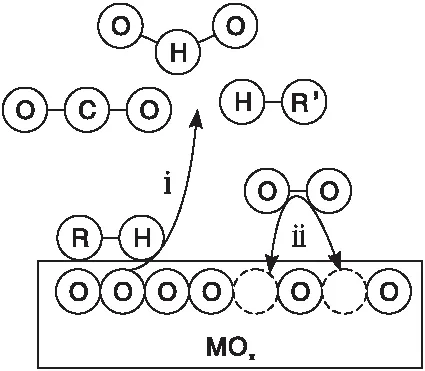
圖1 MVK模型機理Figure 1 The mechanism of MVK model
MVK模型假設VOCs氧化分為兩步進行。首先,被吸附的VOCs與氧氣在催化劑上反應,導致金屬氧化物被還原。其次,氧氣將被還原的金屬氧化物氧化,并繼續留在載體內進行下一步連續循環反應[10]。由于催化劑先被還原后被氧化,這種機制可稱為氧化還原機制。且在穩定狀態下,還原和氧化的速率步驟必須相等。該模型已應用于烴類在催化劑金屬氧化物上的氧化反應動力學模擬研究[11]。根據MVK機制,VOCs氧化速率用式(2)表示。
(2)
式中,rvoc為反應速率,mol·m-3·s-1;Pvoc為揮發性有機化合物的分壓;Po2為氧分壓;Kvoc為揮發性有機化合物氧化速率常數;Ko2為催化劑再氧化速率常數;γ是氧化反應中O2的化學計量系數。
L-H模型機理如圖2所示[12]。
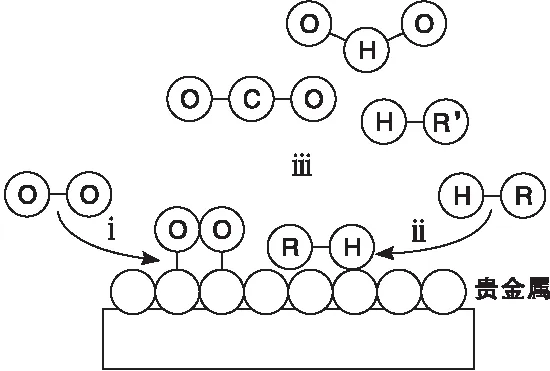
圖2 L-H模型機理Figure 2 The mechanism of L-H model
L-H模型[13]假定吸附在催化劑上的VOCs和氧氣之間發生反應。研究表明,揮發性有機化合物和氧氣可能會吸附在相似類型的活動位點上,被稱為單一位點L-H模型;或是兩個不同類型活動位點上,被稱為競爭位點L-H模型。單一位點L-H模型和競爭位點L-H模型的表達式如式(3)和(4)所示。
(3)
(4)
式中,Ko2為吸附氧平衡常數,Kvoc為吸附VOCs平衡常數。該模型優點是其可以表明反應速率,還可以解釋VOCs和氧的吸附原因。
E-R模型[14]認為,反應是在被吸附的氧物質和氣相中的反應物分子之間發生。其中,吸附的分子和氣相中的分子控制反應步驟。基于E-R模型,式(5)代表其動力學。
(5)
綜上所述,每種機理的合理性很大程度上取決于催化劑(活性金屬和載體)的性質以及揮發性有機化合物的性質。通常狀況下,給定類型的VOCs在特定金屬氧化物或貴金屬催化劑上的反應實驗數據至少會與一種動力學模型機制吻合。
2 催化劑
催化氧化是利用催化劑在較低溫度和較快反應速率等條件下,將待處理有機氣體氧化分解為二氧化碳和水,而且催化劑中負載金屬可以促進有機污染物氧化并減少焦炭形成[15]。所以,工藝關鍵是催化劑的選擇。
2.1 貴金屬催化劑
污染物濃度和性質對反應溫度的選擇具有重要作用,但Pt、Pd、Au及Ag是催化氧化過程中常用的催化劑活性相,這些材料通常會在低溫下對VOCs具有很高的去除率[16-17]。
Pt基催化劑在(150~350) ℃對于VOCs具有較高的去除率[15-18],Pt是氧化芳香族結構最活躍的元素[15-17]。
Pd基催化劑在(200~300) ℃具有較好的催化活性[19-22],而且對于VOCs氣體,Pd基催化劑表現出高的催化活性,比Pt基催化劑更穩定[23]。
Au本身幾乎是化學惰性,但可以增加載體氧化的晶格氧遷移速率[24],與Pd和Pt相比,Au基催化劑在(190~400) ℃對VOCs具有更高的去除率[24-27]。但成本高,限制了含Au催化劑的工業應用,特別是含氯的有機化合物對催化性能也有較大影響[15,28-29]。
使用Ag基催化劑處理VOCs有較多研究,Se-Won Baek等[30]采用XRD對負載Ag的HY沸石進行檢測,發現隨著負載量增加,金屬Ag和Ag2O同時增加,并且負載量與催化活性成正比,表明金屬Ag可能是甲苯氧化的活性相。使用TEM表明,催化活性增加可能與Ag負載量的增加有關。Ye Q等[31]證實了以上結論,確定了納米MnO2中Ag的最佳負載質量分數為5%,沒有證據表明在高負荷條件下催化活性會降低。Kim S C等[32]也得出類似結果,指出γ-Al2O3的最佳Ag負載質量分數為11%。
研究指出,VOCs氧化的最佳條件為Cu、Mn物質的量比等于1,并且在活性相負載質量分數21.2%(Ag、Cu、Mn物質的量比為13.8∶43.1∶43.1)、溫度264 ℃、體積分數0.89%和空速10 000 h-1條件下,對甲苯去除率為最佳[33-34]。數據表明,Ag負載量減少,導致活性中心減少,最終降低了去除效率,但對于更多的負載,活性中心的累積會形成更加低效的整體[35]。
2.2 非貴金屬氧化物催化劑
非貴金屬氧化物比貴金屬便宜,但其催化活性沒有貴金屬好,混合金屬氧化物有可能實現與貴金屬相當的催化效率,所以具有有效催化作用能力的非貴金屬氧化物催化劑經常被應用于去除VOCs。報道較多的是V、Cr、Mn和Ce,其他如Fe、Co和Mo因其催化性能相對較差,報道較少。
使用浸漬法負載質量分數5%~10%的V可制得用于去除氯化VOCs的V基催化劑。在約470 ℃和100 h負載質量分數5.8%的V基催化劑催化活性>90%[36-37]。這種V基催化劑由于其選擇性高,更適合于預處理氯化VOCs。據報道,VOx/TiO2催化劑300 ℃去除約60%的苯(100×10-6,空速37 000 h-1),而且在相同溫度下氧化釩在硫酸化TiO2上轉化率達95%,超過300 ℃轉化率達100%[38]。
Krishnamoorthy S J等[37]研究發現,Cr基催化劑可處理氯化和非氯化VOCs,約280 ℃(600×10-6,空速25 000 h-1)Cr基催化劑可以實現完全去除VOCs中的二氯苯。與V基催化劑相比,起燃溫度高,但可以在較低溫度下進行催化氧化反應[38]。由于其高毒性和潛在的環境問題,限制了實際應用。
使用錳基/TiO2-Al2O3催化劑在350 ℃(1 300×10-6,空速8 000 h-1)下可去除VOCs中97%氯苯,催化活性表現為(10~20) h增加,隨后52 h趨于穩定[38-40]。
報道稱有些非貴金屬氧化物基催化劑顯示出比貴金屬催化劑相當或更好的催化能力[41-42]。含有Zr的Mn基催化劑被證明對VOCs有較好的去除率[43-44]。發現負載于活性炭的Co、Mn混合氧化物比Pd或Au對VOCs中甲苯(10 000×10-6)有更好的去除率,250 ℃時去除率>98%。CoMn/AC氧化物催化劑在250 ℃去除乙苯(10 000×10-6,流速55 mL·min-1)轉化率超過90%,運行周期超過720 h。350 ℃時,這種混合氧化物去除低活性苯不到10%[41]。該差異有可能為不同污染物性質的不同,或者當發生催化氧化反應時,甲苯和乙苯的電離勢能低于苯。
金屬Ce因其優異的儲氧能力是一種不錯的促進劑或載體[45]。被廣泛應用于VOCs的去除,Saqer S M等[46]報道了負載CeO2的氧化鋁催化劑通常需要更高溫才能將污染物去除,且呈現很好的催化活性;同時Cu-Ce和Mn-Ce混合氧化物催化劑在350 ℃能夠將甲苯(1 000×10-6)完全去除。
2.3 雙功能吸附催化劑
雙功能吸附催化劑可以看作是其他類型催化劑既經濟又有效率的替代品,分為兩步法處理VOCs:(1) 預處理:污染物被吸附;(2) 催化氧化:溫度的變化導致污染物解吸、吸附劑再生和有機污染物催化氧化。
制備可控制孔隙大小和比表面積等以增強吸附能力。Brazlauskas M等[47]制備了(100~350) ℃、壽命超過120天可有效氧化醇的雙功能夾層型CuO/沸石吸附劑-催化劑。Kolade M A等[48]制備了高比表面積、低壓降、低阻力和應用于去除VOCs具備高轉化率的負載Pd活性炭吸附催化劑。Roland U等[49]使用Pt-NaY雙功能分子篩催化劑可使VOCs中甲苯完全氧化,催化劑再生溫度為375 ℃。顯然,雙功能催化劑可以發揮各自優勢,為去除VOCs創造更環保、經濟、簡單有效的途徑。
3 工藝流程
催化氧化法處理揮發性有機廢氣凈化效率超過95%,適用于小風量、中高溫度和中高濃度的有機廢氣,且一次投資成本低,運行費用較低,能量回收效果好,符合清潔生產和綠色能源的相關理念。
催化氧化法工藝流程如圖3所示。由圖3可見,經合理收集的VOCs首先通過熱交換器預熱,溫度可升高約(50~200) ℃,然后經過加熱器進一步將VOCs加熱到催化反應溫度約(200~300) ℃,最后,VOCs在催化劑作用下,被轉化成CO2和H2O,同時釋放大量熱能[12]。
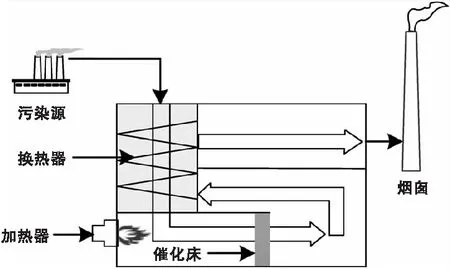
圖3 催化氧化法工藝流程Figure 3 The process for catalytic oxidation
由于熱交換器配置簡化,CO主要節能和優化空間配置。結果表明,CO熱交換效率可達70%,不適合處理大容量、低濃度的VOCs[50-51]。但考慮到初始的低投資和高靈活性,CO適合處理小流量(<5 000 m3·h-1)VOCs。
綜上所述,去除VOCs的核心是使反應溫度降低,即具有低溫和高活性的催化劑。此外,延長催化劑壽命和提高去除效率也可以帶來良好的節能效果和降低投資成本。對于低濃度和大體積的VOCs排放,可通過吸附+催化混合法的先進技術實現去除,已成功應用于實踐[50-52]。
4 結 語
為了深入實施《“十三五”揮發性有機物污染防治工作方案》,打贏藍天保衛戰,提高揮發性有機化合物治理的科學性、針對性和有效性,研究高VOCs治理效率工藝已迫在眉睫。催化氧化技術因其成本低、工藝簡單和處理效率高等優點被廣泛應用,而催化劑開發是其核心,目前,復合金屬氧化物催化劑是研究熱點,因此,結合實際工藝和機理,提高催化劑反應活性和壽命,降低反應溫度和成本等是此領域研究重點。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
光學精密工程(2016年6期)2016-11-07 09:07:19
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
核科學與工程(2015年4期)2015-09-26 11:59:03
