兒童腎透明細胞肉瘤1例
2020-09-18 07:49:12黎美仁王麗麗
臨床與實驗病理學雜志 2020年8期
關鍵詞:兒童
黎美仁,王麗麗,徐 曉,李 婷
患者男性,9歲,3 h前無明顯誘因出現腹痛、腹脹,呈間斷性脹痛,伴惡心、嘔吐,嘔吐胃內容物,無發熱、寒戰。腹部CT:左腎見大小9.2 mm×9.8 cm×15 cm團塊狀占位,病灶邊緣清楚,由左腎動脈供血,增強掃描實性成分呈輕度強化,左腎殘余腎實質未見異常強化,周圍組織受推擠移位。遂行左腎根治術并送檢。
病理檢查眼觀:腎臟切除標本1個,大小13 cm×5 cm×4 cm,腎被膜尚完整,附部分輸尿管,長12 cm,管徑0.8~1.5 cm,腎臟一側可見腫物1個,大小15 cm×12 cm×10 cm,腫物累犯整個腎臟,切面實性,局灶可見囊腔形成,囊內含淡黃色清亮液體,切面灰白、灰黃色,魚肉狀,局灶出血,質軟。鏡檢:瘤細胞彌漫增生,呈束狀、條索狀或巢團狀,細胞呈短梭形、卵圓形或多角形,細胞大小較一致,核染色質細膩,核仁不明顯,核分裂象易見(20個/10 HPF),胞質淡染、透亮,可見小灶壞死,間質薄壁小血管豐富(圖1、2)。免疫表型:vimentin呈彌漫強陽性(圖3),SATB2呈彌漫陽性(圖4),BCOR呈弱陽性(圖5),CAⅨ、Cyclin D1、CD10、CD117、Pax-2、Pax-8均陽性,其它標記CK(AE1/AE3)、BCL-2、CK7、desmin、E-cadherin、P504s、NSE、WT-1及HMB45均呈陰性,Ki-67增殖指數約40%。PCR檢測結果顯示BCOR基因15號外顯子ITD存在串聯重復(圖6)。
病理診斷:(左腎切除標本)腎透明細胞肉瘤(clear cell sarcoma of the kidney, CCSK)。
討論CCSK是除Wilms瘤外最常見的兒童腎臟惡性腫瘤,因其臨床表現及發病年齡類似Wilms瘤,曾被認為是它的一種特殊亞型。CCSK約占兒童腎臟惡性腫瘤的3%,男女比約為2 ∶1,多發生在3歲以下兒童。患者常見臨床癥狀主要有腹痛、腹脹、腹部包塊以及肉眼血尿,發生骨轉移時可出現骨痛。
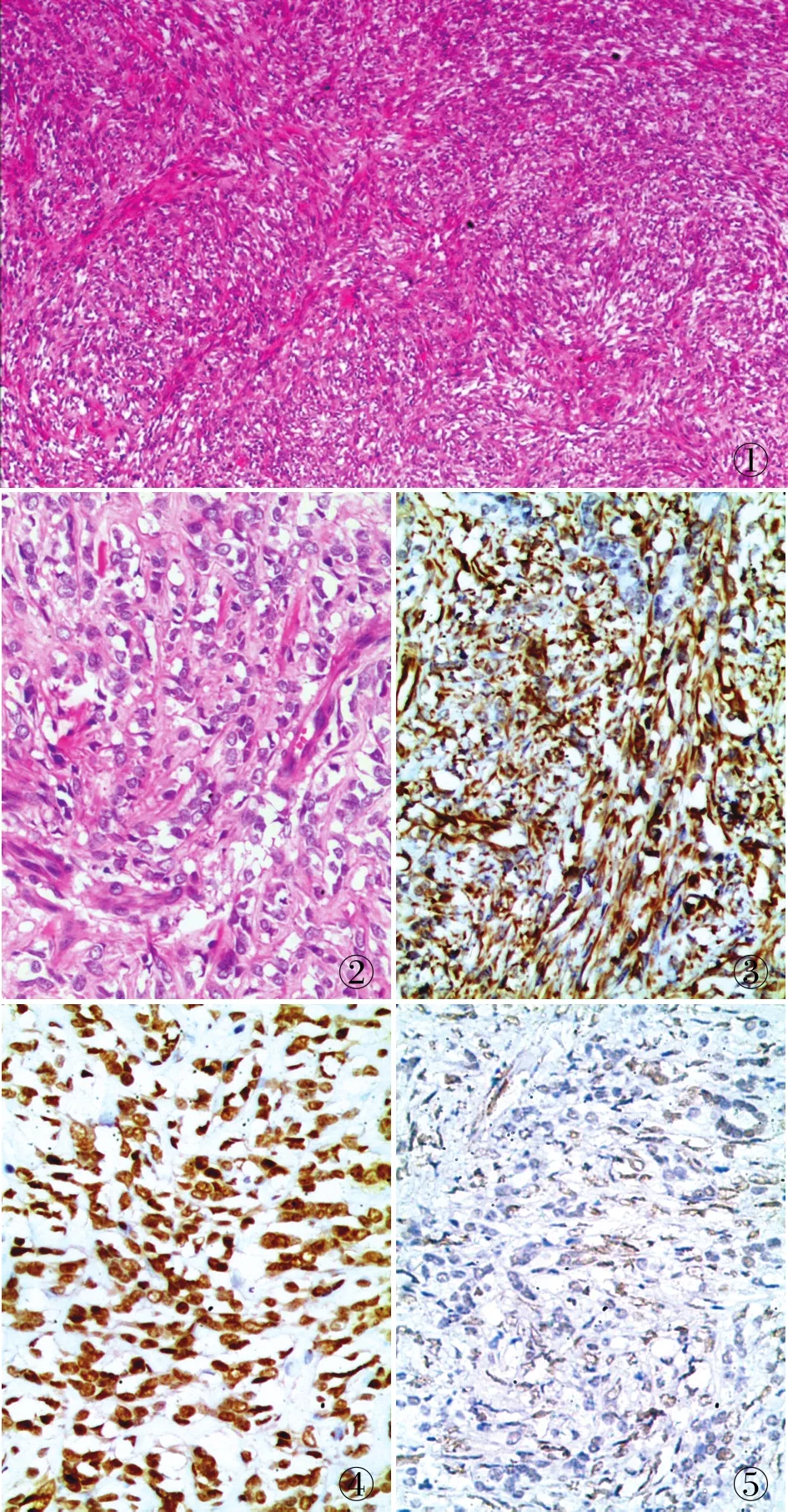
①②③④⑤
CCSK體積一般較大,平均直徑11 cm,好發于腎髓質或腎中央,單發,界清,無包膜,切面魚肉狀,黏液樣,多數會有囊性變。鏡下瘤細胞呈多邊形,核染色質細膩,核仁不明顯,胞質含有多數透明的空泡,呈巢索狀排列,腫瘤間可見豐富的網狀毛細血管。腫瘤細胞中vimentin、Cyclin D1、SATB2、TLE1、CD10、BCL-2均陽性,而CD34、desmin、CD99、S-100及上皮源性標記均陰性[1]。有研究發現,CCSK中BCOR蛋白的陽性率為100%(20/20)[2],其敏感性及特異性高。在分子遺傳學方面,CCSK主要存在BCOR基因中15號外顯子ITD串聯復制和YWHAE-NUTM2B/E基因融合,前者檢出率約85%,后者約10%,目前尚未有單一病例同時檢出的報道,提示兩者可能存在互斥性[3]。

圖6 PCR檢測示BCOR基因15號外顯子ITD存在串聯重復
鑒別診斷:(1)Wilms瘤:由未分化腎胚芽組織及多少不等的上皮、間葉成分構成,與CCSK形態上有交叉,但CCSK無腎胚芽成分,與正常腎組織分界不清,WT-1、CK均陰性。(2)兒童惡性橫紋樣腫瘤(rhabdoid tumour of kidney, RTK):好發于兒童腎或中樞系統,主要特點為囊泡狀染色質、核仁明顯及胞質內玻璃樣變的包涵體,INI-1陰性,而CCSK腫瘤細胞核染色細膩,無紅染的胞質或核內包涵體,“雞爪樣”血管豐富,無INI-1蛋白缺失。(3)軟組織透明細胞肉瘤:俗稱“軟組織惡性黑色素瘤”,腫瘤細胞質透亮,但表達S-100及HMB-45,無黑色素沉積,電鏡下可見黑色素小體,較少發生于兒童,雖然都稱為透明細胞肉瘤,但與CCSK的生長模式及免疫表型差異較大,易于鑒別。(4)先天性中胚層腎瘤(congenital mesoblastic nephroma, CMN):患者多于出生后6個月內發病,1歲后發病者罕見,主要由交錯排列的纖維母細胞構成,細胞溫和,核分裂象少見,細胞無明顯的分化方向,間質可見“鹿角樣”血管,與CCSK鑒別的主要為細胞型CMN,腫瘤細胞豐富,呈條索或片狀,核分裂象及壞死常見,腫瘤一般表達vimentin及actin。(5)孤立性纖維性腫瘤(solitary fibrous tumour, SFT):腫瘤細胞呈血管周細胞瘤樣增生模式,密集區及稀疏區交錯排列,CD34、STAT6、BCL-2及CD99均陽性可資鑒別。最近研究顯示,發生在腎臟的惡性SFT會出現BCOR蛋白強陽性,但不出現BCOR基因異常,所以診斷CCSK時,不能僅因BCOR蛋白陽性就直接診斷,需結合HE形態和其他免疫組化特點,必要時還需進行相關基因檢測[4]。
目前,CCSK發病機制尚不清楚,有學者[5]認為其發生與TCF21基因甲基化相關,TCF21屬于抑癌基因,又稱Pod-1、Capsulin或Epicardin,位于6q23,是一種已知在腎臟發育早期具有活性的轉錄因子。在該研究中,絕大多數CCSK(12/13)病例中均存在抑癌基因TCF21啟動子的甲基化以及蛋白的低表達。同時,多數病例中也未檢測到TARID,它是一種非編碼長鏈RNA,其主要功能是對TCF21基因去甲基化。TCF21高甲基化和TARID表達下降在其他CCSK獨立樣本中也得到驗證。有學者提出:TCF21高甲基化和(或)TARID表達降低,可能是CCSK發病的重要因素,但其與腫瘤特有的基因表達模式間的關系有待進一步分析。
腫瘤治療一般以手術切除為主,并聯合術后放、化療,即使不能完整切除腫瘤時,也建議盡可能行減瘤術,淋巴結清掃不能改善預后,但有助于對腫瘤進行分期,腫瘤診斷時腎周淋巴結轉移率高達29%。患者在接受綜合性治療后,5年總生存率為85%~90%[6]。CCSK極易發生骨轉移,發生率為40%~60%,其中以顱骨居多。腫瘤性壞死的出現可能降低生存期[7],發生在成人時預后更差[8]。
(本例承蒙解放軍東部戰區醫院病理科饒秋教授指導,特此致謝!)
猜你喜歡
少兒美術·書法版(2021年12期)2021-10-24 02:50:16
少兒美術·書法版(2021年9期)2021-10-20 06:35:28
少兒美術·書法版(2021年7期)2021-10-20 06:29:16
少兒美術·書法版(2021年11期)2021-10-20 06:23:28
少兒美術·書法版(2021年10期)2021-10-20 06:14:04
少兒美術·書法版(2021年8期)2021-10-20 06:08:10
少兒美術(2019年8期)2019-12-14 08:07:00
少兒美術(2019年3期)2019-12-14 08:02:56
雜文選刊(2016年7期)2016-08-02 08:39:56
小天使·一年級語數英綜合(2016年6期)2016-05-14 12:21:05
