“甘草-馬錢子”配伍前后及其拆分相態指紋圖譜對比研究
2020-10-30 13:13:52郭玉巖李春成孫爽楊大宇呂邵娃
中醫藥學報 2020年10期
郭玉巖,李春成,孫爽,楊大宇,呂邵娃
(黑龍江中醫藥大學藥學院,黑龍江 哈爾濱 150040)
中藥配伍減毒是中醫藥學的特色和優勢之一,有毒中藥通過與其它藥物配伍,不僅可以增強藥力、提高療效,同時又可減輕或消除毒副作用。馬錢子為典型的有毒中藥,其因止痛作用較為明顯[1],為歷代醫家所推崇,與甘草配伍為其常見的減毒方法[2],古籍有記“生馬錢子加甘草浸泡”“服馬錢子量大時,飲甘草水解之”“馬錢子內服方劑與甘草配伍”或“甘草炙馬錢子”等,現代含有馬錢子的舒筋丸、疏風定痛丸等口服制劑處方也均見與甘草配伍使用,足見甘草能緩和馬錢子的毒性。目前已有不少學者關注了甘草配伍馬錢子減毒的這一現象,本課題組前期研究發現,甘草和馬錢子在水煎煮過程中生成了大量沉淀,推測煎煮過程中溶入水體的次生代謝小分子成分、生物大分子物質以及無機元素等均影響和改變著湯液的理化性質,這些天然的表面活性劑,可起到助懸、絮凝和反絮凝等多種作用,使得湯液成為真溶液、膠體溶液和混懸液及大分子沉淀顆粒同時存在的復雜分散相態;進一步對各相態中的毒效物質含量進行測定發現,沉積物相態富集了近80%的毒效物[3]。然而甘草與馬錢子配伍前后是否有化學成分的改變,不同產地、批次藥材配伍后化學成分的監控,以及甘草-馬錢子配伍拆分相態、化學成分的迥異均仍有待進一步研究。
指紋圖譜技術是能夠標示中藥化學成分特征的經典技術,其既可以對已知成分進行分析,也可以對未知成分進行分析[4],可較為全面地反映混合的復雜體系中各種化學成分及其含量情況[5],為此,本文采用指紋圖譜技術,對甘草-馬錢子藥對配伍前后及其拆分相態的化學成分進行特征分析,以期為甘草馬錢子的配伍應用提供檢測手段,并為甘草解馬錢子之毒的研究提供參考。
1 儀器與材料
1.1 儀器
UItiMate3000色譜儀(美國Thermo Fisher Scienti-fic公司);AB265-S分析電子天平(美國METTLER-TOLEDO公司);SB-5200D型超聲波清洗機(寧波新芝生物科技股份有限公司)。
1.2 材料
馬錢子、甘草藥材購自于北京同仁堂哈爾濱藥店有限責任公司;馬錢子堿(Y16A7S13272)、士的寧(Z02A7S18869)均購自上海源葉生物科技有限公司;甘草苷(HL04223898)、甘草素(HL042236198)、甘草酸(HI042238196)、異甘草素(HL042236198)、甘草查爾酮A(HL042237198)、甘草次酸(HL042902198)、異甘草苷(HI042234198)、芒柄花素(HL042235198) 均購自寶雞市晨光科技有限公司,甲醇、乙腈、冰醋酸、濃鹽酸、 三乙胺、磷酸等均為分析純級試劑。
2 方法與結果
2.1 色譜條件
采用Diamonsil C18(2)(250 mm×4.6 mm 5 μm)色譜柱;以乙腈(B)-0.1%磷酸水(A)為流動相,梯度洗脫(0~10 min,5%~10% A;10~20 min,10%~13% A;20~30 min,13%~15% A;30~40 min,15%~20%A;40~45 min,20%~23%A,45~60 min,23%~32%A,60~80 min,32%~38%A;80~90 min,38%~48%A;90~105 min,48%~65%A;100~115 min,80%A);流速1 mL/min;柱溫25 ℃;檢測波長254 nm;進樣量10 μL。
2.2 對照品溶液的制備
取士的寧、馬錢子堿標準品適量,置10 mL量瓶中,加甲醇超聲溶解,定容至刻度,使濃度分別0.205、0.215 mg/mL。另取甘草苷、異甘草苷、甘草酸、甘草素、甘草查爾酮A、異甘草素、甘草次酸、芒柄花素對照品適量,置10 mL量瓶中,加甲醇超聲溶解,定容至刻度,制成濃度分別為0.189、0.198、0.250、0.216、0.202、0.246、0.217 mg/mL的對照品溶液。
2.3 供試品溶液的制備
2.3.1 甘草、馬錢子及甘草-馬錢子配伍供試品溶液的制備
分別稱取0.75 g甘草,0.25 g馬錢子,0.75 g甘草與0.25 g馬錢子的混合粉末,分別置于25 mL量瓶中,加鹽酸-甲醇-水(1∶50∶50)適量,超聲處理45 min,放冷,用鹽酸-甲醇-水(1∶50∶50)補足至刻度,搖勻,過濾,取續濾液即分別為甘草、馬錢子及二者配伍供試品溶液。
2.3.2 甘草-馬錢子配伍相態供試品溶液的制備
按文獻進行湯液相態拆分:精密移取馬錢子甘草共煎液,采用低溫沉降,高速離心以及生物膜透析的方法將湯液拆分成馬錢子甘草共煎液沉積物組、混懸物組、膠體液分、溶液組4個相態部分[6]。所有樣品真空冷凍干燥24 h,取出備用。
拆分相態供試品溶液的制備:分別取馬錢子甘草湯液相態凍干粉約1 g,加鹽酸-甲醇-水(1∶50∶50)適量,超聲處理45 min,放冷,用鹽酸-甲醇-水(1∶50∶50)補足至刻度,搖勻,過濾,取續濾液即得甘草-馬錢子相態的供試品溶液。
2.4 方法學考察
2.4.1 精密度試驗
取甘草-馬錢子配伍供試品,按“2.3”項下的方法制備供試品溶液,按“2.1” 項下的的色譜條件連續進樣6次,分別以甘草酸的色譜峰為參照峰(S),測定并計算得到共有峰相對保留時間RSD<2%,表明儀器精密度良好。
2.4.2 穩定性試驗
取甘草-馬錢子配伍供試品,按“2.3”項下的方法制備供試品溶液,按“2.1” 項下的的色譜條件,分別于0,2,4,6,8,12 h進樣測定以甘草酸為參照峰(S),測定并計算得到共有峰相對保留時間RSD<2%,表明穩定性良好。
2.4.3 重復性試驗
取甘草-馬錢子配伍供試品,按“2.3”項下的方法分別平行制備6份供試品溶液,按“2.1” 項下的的色譜條件進樣6次,以甘草酸的色譜峰為參照峰(S),測定并計算得到共有峰相對保留時間RSD<2%,表明重復性良好。
2.5 指紋圖譜及相似性評價
按“2.3”項下的方法,分別制備甘草、馬錢子,甘草-馬錢子配伍供試品溶液及四種相態拆分樣品溶液。按照“2.1”項下色譜條件進樣測定并記錄UPLC 色譜圖。將甘草-馬錢子配伍供試品溶液色譜圖cdf文件導入國家藥典委員會“中藥色譜指紋圖譜相似度評價系統(2012版)”中,然后進行譜峰匹配, 通過與對照品比對,共確定25個共有峰。指認出9個成分。結果見圖1、圖2,表1、表2。通過比較甘草-馬錢子配伍供試品溶及混合對照品的圖譜,對合煎液中各共有峰進行初步歸屬,1~5峰歸屬于馬錢子,6~25峰歸屬于甘草。通過保留時間和 UV 光譜的對照,確定色譜峰4,5,8,11,12,16,18,23,25,分別對應馬錢子堿、士的寧、甘草苷、異甘草苷、甘草素、異甘草素、甘草酸、甘草查爾酮A、甘草次酸9個指標性成分;分別計算各共有峰的相對保留時間RSD為0.31%~2.80%。不同樣品各共有峰的峰面積相差較大,表明不同產地甘草與馬錢子的化學成分含量存在一定的差異。配伍后色譜與對照圖譜的相似度均在0.900以上,說明樣品指紋圖譜與對照圖譜的相似度較高,符合指紋圖譜的要求。
2.6 配伍前后指紋圖譜對比分析
通過對馬錢子、甘草單煎及其配伍前后指紋圖譜對比分析,鑒定結果顯示,經過配伍后,經過配伍后,沒有新峰出現,沒有出現差異峰,而經配伍后士的寧與甘草酸的峰面積顯著減少,推測甘草-馬錢子配伍后減毒可能因為物理變化,結果見圖3。
2.7 不同相態指紋圖譜對比分析
以9種指標性成分對馬錢子甘草配伍拆分后的四種相態色譜圖進行對比分析發現,甘草-馬錢子配伍后的活性成分大多數集中在沉積物相態中,在四種相態中,均有4、5、8、11號峰,分別為馬錢子堿、士的寧、甘草苷、異甘草苷。均檢是馬錢子-甘草配伍相態溶液中的特征性成分。其中D相態中包含所有指認峰,A、B相態均有4、5、8、11、16、18,分別為馬錢子堿、士的寧、甘草苷、異甘草苷、異甘草素、甘草酸。C相態中指標性成分最少。
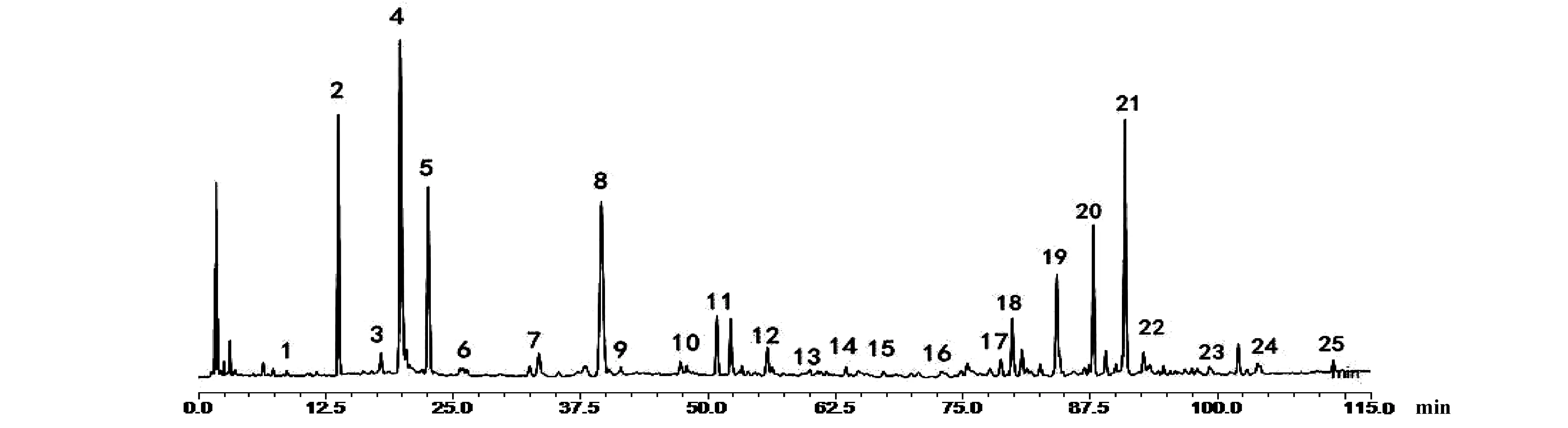
注:4.馬錢子堿;5.士的寧;8.甘草苷;11.異甘草苷;12.甘草酸;17.甘草素;19.異甘草素;22.甘草查爾酮A;24.甘草次酸。圖1 馬錢子甘草配伍樣品UPLC圖譜
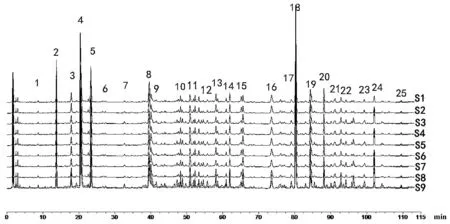
圖2 馬錢子甘草配伍樣品UPLC疊加圖譜
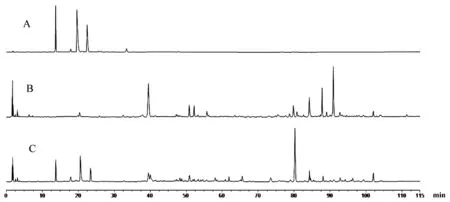
注:A.馬錢子供試品;B.甘草供試品;C.馬錢子-甘草配伍供試品。圖3 馬錢子-甘草配伍前后指紋圖譜對比UPLC色譜圖
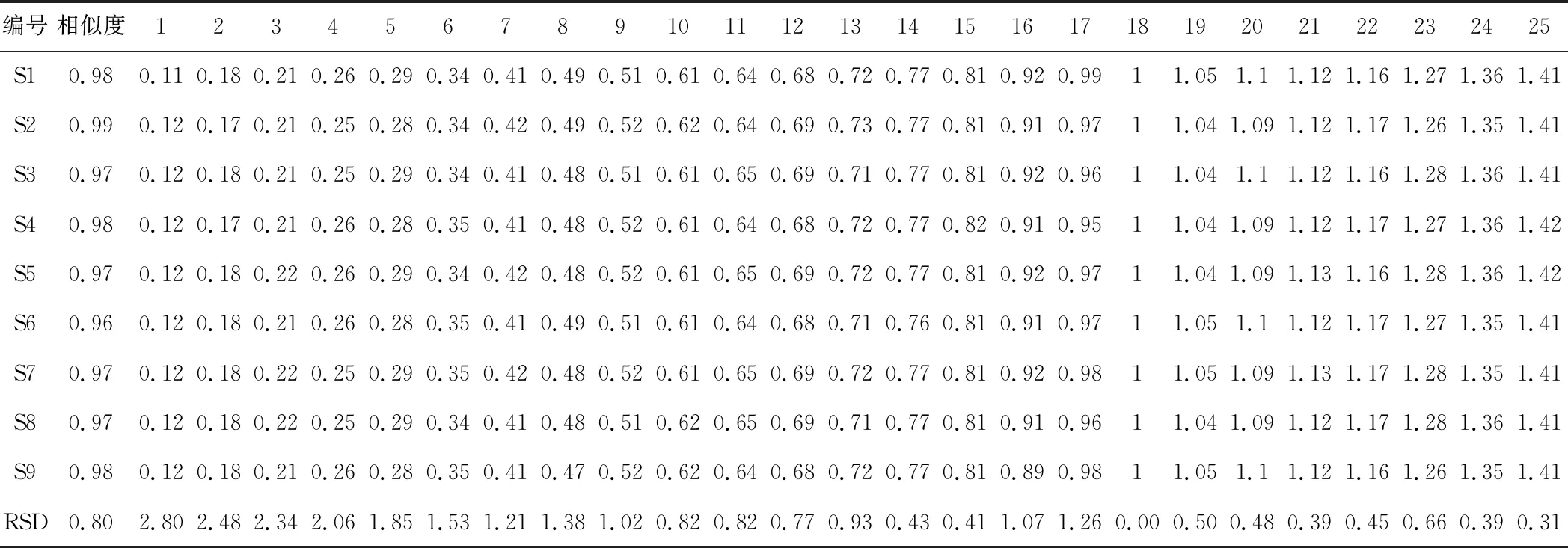
表1 馬錢子-甘草配伍樣品共有峰的相對保留時間及相似度
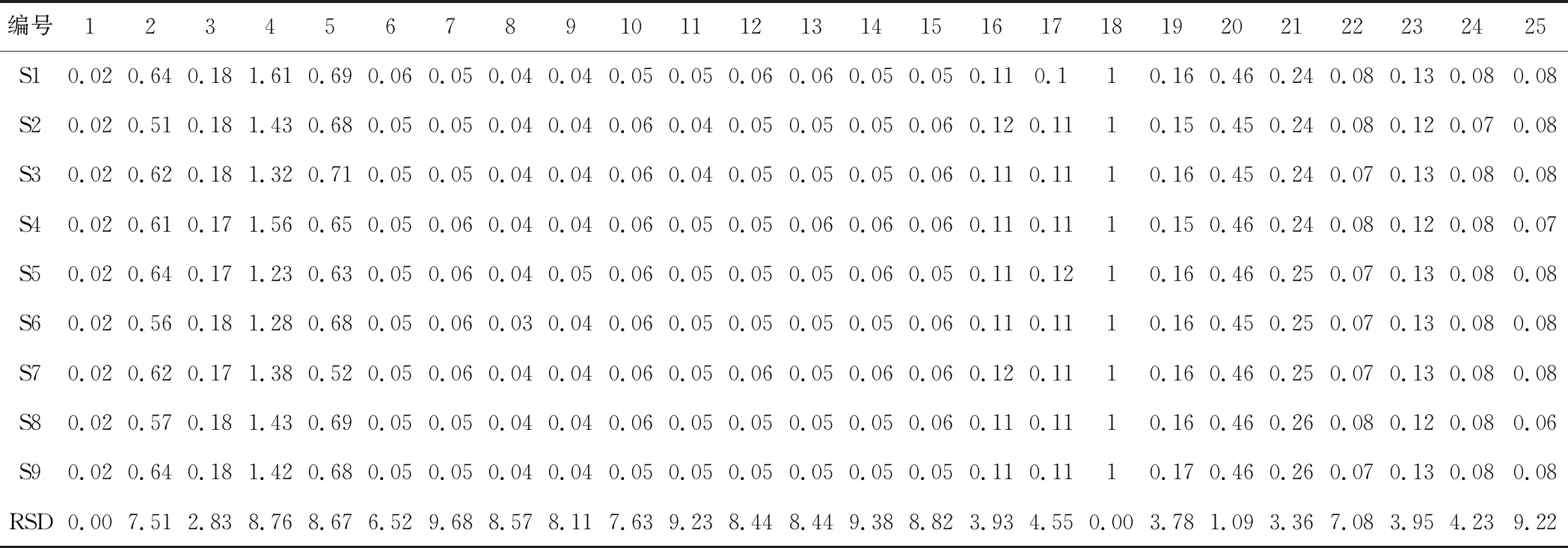
表2 馬錢子-甘草配伍樣品共有峰的相對峰面積
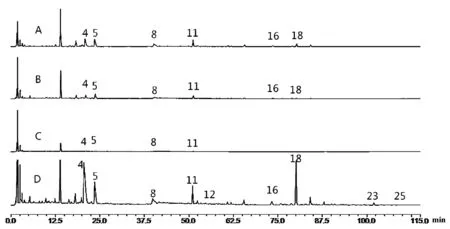
注:A.混懸相態;B.膠體相態;C.溶液相態;D.沉淀相態。圖4 馬錢子-甘草配伍四種相態的UPLC色譜圖
3 討論
3.1 提取溶劑的選擇
考察以水、甲醇-水(1∶1)、鹽酸-甲醇-水(1∶50∶50)為溶劑對馬錢子、甘草單煎液、馬錢子-甘草配伍液的指紋圖譜的影響[7]。結果表明,以鹽酸-甲醇-水(1∶50∶50)為溶劑,超聲提取提取所得的色譜峰信息量相當,綜合考慮溶劑提取能力、色譜結果的準確性,確定以鹽酸-甲醇-水(1∶50∶50)為超聲提取制備供試品。
3.2 流動相的選擇
分別考察乙腈-1%乙酸、乙腈-0.2%乙酸-0.2%三乙胺、乙腈-0.2%乙酸、乙腈-甲酸水(pH=2.4)及乙腈-0.1%磷酸水為流動相,并不斷調整流動相的比例來確定甘草-馬錢子最佳色譜條件[8]。結果以乙腈-0.1%磷酸水為流動相,在選定的條件下,峰信息量較多,各色譜峰的分離效果較好,峰形優良。
3.3 馬錢子-甘草指紋圖譜的建立
甘草-馬錢子是經典的苦甘藥對,甘草對馬錢子減毒增效作用顯著,是臨床上常見配伍藥對。目前已有金永新等對烏拉爾甘草進行高效液相指紋圖譜的研究[9]。王月輝、喬菲等[10-11]等采用HPLC進行了馬錢子指紋圖譜的研究,建立了指紋圖譜,具有重要的現實意義。然而,由于毒性限制,馬錢子在臨床中很少單獨使用,上述研究立足于甘草與馬錢子單獨的指紋圖譜研究,卻忽略了甘草與馬錢子配伍前后化學成分的變化和復方中各個成分間作用的“復雜性”“整體性”,難以較好詮釋兩者配伍“毒解而效不減的作用機制。本研究建立馬錢子-甘草配伍后的指紋圖譜方法,指認了9個共有峰,相似度評價結果顯示,該結直觀、可靠、可操作性強,可為甘草和馬錢子的配伍應用提供質量研究方法。
3.4 馬錢子-甘草配伍前后化學成分的改變
中藥復方煎煮過程中常伴隨一系列復雜的物理、化學反應,通過對關鍵藥對的分析,可深入挖掘中藥復方配伍規律,化學成分是藥對產生療效的物質基礎,因此可以從藥對配伍前后的化學成分變化著手探究配伍基礎。由于現階段多數中藥物質基礎不明確,指紋圖譜可更全面反應藥對所含化學成分的種類與數量,可進一步闡明藥對配伍增效的物質變化基礎,體現了配伍過程中化學成分的整體變化情況。本文在指紋圖譜方法的基礎上,對馬錢子-甘草配伍前后的色譜峰進行對比研究,結果顯示與單煎液相比,配伍后的色譜峰個數并沒有明顯變化,沒有新峰出現;然而,配伍樣品的色譜峰可見士的寧與甘草酸的峰面積顯著降低,且本課題組對配伍湯液進行拆分時發現,其有大量新生沉淀的產生,推測馬錢子和甘草合煎過程,可能存在分子絡合等物理反應的發生而起到減毒增效的作用。
3.5 馬錢子-甘草配伍湯液相態的色譜圖對比分析
近年來,以相態角度對有毒中藥的研究已取得一定的成果,例如研究發現附子-甘草藥對配伍時,其合煎渾濁湯液靜置后出現大量沉積物[12],又有研究者發現,部分藥對如黃芩-黃連合煎產生的沉淀影響藥物效應等[13]。本課題組前期[3]將甘草-馬錢子進行了相態拆分,獲得溶液相態、膠體相態、混懸相態和沉積物4個相態,并對毒效成分的相態分布進行了研究,本文通過指紋圖譜方法對4個相態的成分進行對比分析,結果表明沉積物相態為活性成分的聚集相態,該相態的進一步深入解析,可為馬錢子甘草配伍減毒機制提供又一突破。
