麻黃含量測定方法研究
2021-01-25 11:57:34肖丹丹孫瑩瑩徐自信劉芳馨林喆李勇
世界最新醫(yī)學(xué)信息文摘 2020年95期
肖丹丹,孫瑩瑩,徐自信,劉芳馨,林喆,李勇
(長春中醫(yī)藥大學(xué)藥學(xué)院,吉林 長春)
0 引言
麻黃(Herba Ephedrae)為多基原傳統(tǒng)中藥(中麻黃、草麻黃及木賊麻黃),始載于《神農(nóng)本草經(jīng)》,列為中品。其性溫、味辛、微苦;歸肺、膀胱經(jīng);具有發(fā)汗散寒[1]、宣肺平喘、利水消腫及治療腹膜炎等多種功效,僅2015版《藥典》收載含有麻黃的成方制劑就多達56個。現(xiàn)代藥理學(xué)研究表明其主要藥效物質(zhì)基礎(chǔ)為麻黃堿與偽麻黃堿,2005年藥典即對所含麻黃堿做了明確的限量規(guī)定,而2010版和現(xiàn)行2015版藥典又增加了偽麻黃堿的含量測定,可見準(zhǔn)確、穩(wěn)定、可行的含測方法尤為重要。
文獻研究表明,歷版藥典[2-4]用于麻黃含測的高效液相色譜法(HPLC)在不斷完善,即從反相C18鍵合硅膠柱(2005版,流動相:ACN-0.1% H3PO3,9:87)變?yōu)闃O性乙醚連接苯基鍵合硅膠柱(2010版,流動相:MeOH-含0.04%三乙胺與0.02%二正丁胺的H3PO3溶液,1.5:98.5;2015版,流動相:MeOH-含0.04%三乙胺與0.02%二正丁胺的0.092%H3PO3溶液,1.5:98.5),其目的是能夠同時準(zhǔn)確檢測上述兩種立體異構(gòu)體(麻黃堿與偽麻黃堿)。但由于色譜條件變化后的要求越來越高,致使應(yīng)用并不普遍,如2015版藥典所載以麻黃為含測指標(biāo)的22個中成藥中,僅有2味制劑選用極性乙醚柱及相應(yīng)色譜條件,其他均為C18色譜柱;另據(jù)文獻檢索顯示,自2010版藥典改用極性乙醚色譜柱至今的10年間,中國知網(wǎng)、維普、清華同方、萬方及超星數(shù)據(jù)庫中報導(dǎo)使用極性乙醚柱的文獻僅8篇[5-12],其中含有色譜圖的僅4篇。
本文采用常規(guī)普遍使用的C18色譜柱,針對其主要影響因素的流動相進行了初步的優(yōu)化研究,同時結(jié)合相關(guān)橫向課題(校企橫向課題,吉林省利華制藥有限公司),以優(yōu)化后的色譜條件對中麻黃飲片進行了實際的含量測定研究,結(jié)果表明該方法準(zhǔn)確、重現(xiàn)性好且易于操作,為麻黃含測方法的進一步完善提供了實驗依據(jù)。
1 材料
1.1 儀器
高效液相色譜儀(SPD-10A VPLC-10AT VPAT-330,日本島津);XS205型電子分析天平(梅特勒-托利多儀器有限公司);ST-2000旋轉(zhuǎn)蒸發(fā)儀(上海亞榮生化儀器廠);超聲波清洗器(型號:KQ200B,昆山市超聲儀器有限公司)。
1.2 試藥與試劑
鹽酸麻黃堿對照品(供含量測定用,批號171241-201809)、鹽酸偽麻黃堿對照品(供含量測定用,批號171237-201510)均購自中國食品藥品檢定研究院。乙腈、磷酸為色譜純;其他試劑均為分析純。
1.3 樣品收集
購于吉林大藥房,經(jīng)長春中醫(yī)藥大學(xué)林喆教授鑒定為中麻黃(Ephedra intermedia Schrenk et C. A. Mey.)。
2 方法與結(jié)果
2.1 溶液的制備
2.1.1 對照品溶液的制備
取鹽酸麻黃堿對照品、鹽酸偽麻黃堿對照品適量,精密稱定,加50%甲醇溶液分別制成每l mL含40 μg的溶液,即得。
2.1.2 供試品溶液制備
精密稱取麻黃藥材粉末(過3號篩)1 g,置具塞錐形瓶中,精密加入50%甲醇溶液100 mL,密塞,超聲處理(功率為250 W,頻率為50 Hz)30 min,放冷,再稱定重量,用50%甲醇溶液補足減失的重量,搖勻,濾過,取續(xù)濾液即得。
2.2 色譜條件優(yōu)化
在采用極性乙醚色譜柱且有色譜圖的文獻中可見(僅4篇),無論采用2010版藥典流動相(日本島津公司報道見圖1A)還是2015版流動相(見圖1B)均未獲得最佳分離色譜,其中前者鹽酸麻黃堿有嚴(yán)重的拖尾現(xiàn)象;而后者鹽酸麻黃堿峰對稱性不佳,直接影響了含量測定的準(zhǔn)確性。
筆者采用2005年版藥典方法對鹽酸麻黃堿和鹽酸偽麻黃堿對照品及麻黃藥材分別進行了重復(fù)試驗,對照品及藥材中兩個成分均未達到基線分離(見圖1C);另外依據(jù)最新文獻報道色譜條件,即 C18 色譜柱(4.6 mm ×250 mm,5 μm)、乙腈-0.2%磷酸溶液(4:96)為流動相、檢測波長為210 nm、柱溫25 ℃及流速0.8 mL/min進行了重復(fù)實驗,結(jié)果顯示鹽酸偽麻黃堿與甲基麻黃堿并未完全分離(見圖1D)。
經(jīng)對所有色譜條件反復(fù)多次的考察,發(fā)現(xiàn)流速、柱溫及檢測波長并非主要影響因素,因此對流動相進行了系列梯度變化摸索研究,最終確定流動相為乙腈-0.2%磷酸溶液(1.5:98.5),兩峰均得到完全分離(見圖2A),兩峰理論塔板數(shù)和拖尾因子均符合現(xiàn)行版藥典要求。故確定色譜最佳條件:上述流動相、C18色譜柱(4.6 mm×250 mm,5 μm)、檢測波長為 210 nm,柱溫 25 ℃,流速 0.8 mL/min。
2.3 方法學(xué)考察
2.3.1 系統(tǒng)適用性試驗
取上述對照品溶液和供試品溶液適量,按“2.2”項下色譜條件進樣測定,記錄色譜圖,見圖2。鹽酸麻黃堿標(biāo)準(zhǔn)品液相色譜圖中苦參堿保留時間為57 min,鹽酸偽麻黃堿保留時間69 min,理論板數(shù)均>3000。
2.3.2 線性關(guān)系考察
分別精密稱取鹽酸麻黃堿、鹽酸偽麻黃堿對照品2.19 mg和2.18 mg,置10 mL容量瓶中,用50%甲醇溶液溶解并稀釋至刻度,搖勻,分別精密量取 0.5、1.0、1.5、2.0、2.5 mL,置 10 mL 容量瓶中,用50%甲醇溶液溶解并稀釋至刻度,搖勻,作為測試溶液,分別精密量取各溶液10 μL注入液相色譜儀,記錄色譜圖,以峰面積對相應(yīng)濃度進行線性回歸處理。結(jié)果表明:鹽酸麻黃堿在10.90~180.00 μg/mL線性關(guān)系良好,回歸方程為Y=9701.7X+847,r2=0.9994;鹽酸偽麻黃堿在 10.95~80.00 μg/mL 線性關(guān)系良好,回歸方程為Y=9766.5X-750.7,r2=0.9995。

圖1 藥典(2010、2015和2005版)和參考文獻方法HPLC色譜
2.3.3 精密度實驗
取“2.1.1”項下對照品溶液,按“2.2”項下色譜條件連續(xù)進樣6次,測定峰面積。測得RSD=1.79%,表明儀器精密度良好。
2.3.4 重復(fù)性實驗
取麻黃樣品約1 g,按照“2.1.2”下供試品溶液制備方法制備5份,按“2.2”項下色譜條件。進行含量測定,計算RSD值為1.66%,表明樣品處理方法重復(fù)性良好。
2.3.5 穩(wěn)定性實驗
按“2.1.1”項下對照品的制備方法,制備新對照溶液,按“2.1”項下色譜條件連續(xù)注入液相色譜分別于 0、2、4、6、8、10、12 h的標(biāo)準(zhǔn)品并測定峰面積,鹽酸麻黃堿、鹽酸偽麻黃堿峰RSD值分別為1.68%和1.45%,表明溶液在12 h內(nèi)穩(wěn)定。
2.3.6 加樣回收率實驗
取的麻黃樣品1.0 g共18份,分兩大組,兩大組分三小組分別精密加入鹽酸麻黃堿、鹽酸偽麻黃堿標(biāo)準(zhǔn)品相當(dāng)于樣品濃度的80%、100%、120%。按照制備“2.1.2”項下供試品溶液的提取方法提取,另取“2.1.1”項下對照品溶液進樣測定做對照,并計算鹽酸麻黃堿、鹽酸偽麻黃堿低中高3個濃度的加樣回收率,結(jié)果見表1。

圖2 麻黃中鹽酸麻黃堿、鹽酸偽麻黃堿HPLC色譜圖

表1 鹽酸麻黃堿、鹽酸偽麻黃堿加樣回收率試驗結(jié)果(n=9)
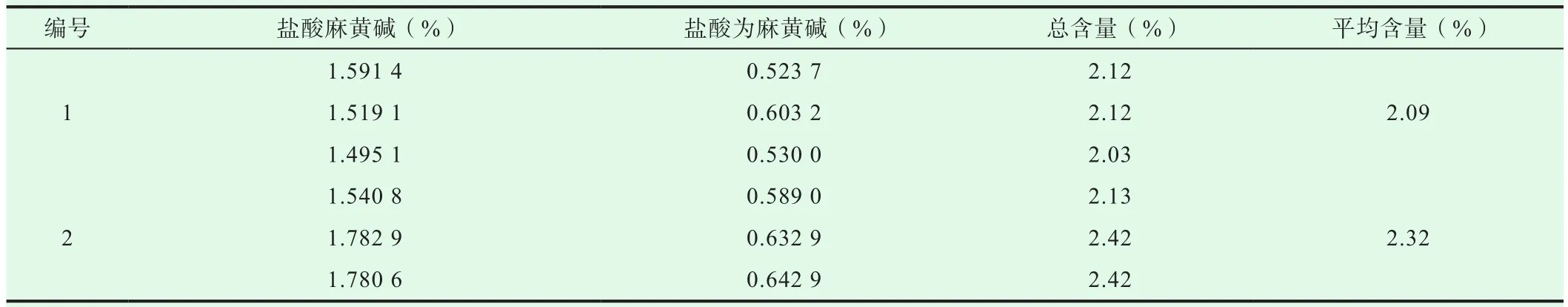
表2 麻黃含量測定(n=3)
結(jié)果表明,鹽酸麻黃堿在低中高3個濃度中的加樣回收率均在98.7%~101.8%,平均回收率100.39%,RSD值為1.18%,鹽酸偽麻黃堿在低中高3個濃度中的加樣回收率均在98.4%~101.8%,平均回收率100.11%,RSD值為1.16%,表明本方法回收率良好。
2.4 含量測定
取麻黃樣品,粉碎成粉末,過三號篩。精密稱取1.0 g,各3份,按“2.1.2”項下方法制備供試品溶液。另取“2.1.1”項下對照品溶液作對照,按“2.2”項下色譜條件進行樣品測定,計算麻黃供試品的中鹽酸麻黃堿、鹽酸偽麻黃堿含量。所測樣品的鹽酸麻黃堿和鹽酸偽麻黃堿含量均>0.80%,結(jié)果符合藥典規(guī)定結(jié)果,見表2。
3 討論
3.1 色譜柱
建議繼續(xù)使用常規(guī)普遍應(yīng)用的C18反相色譜柱。雖然極性乙醚色譜柱具有更好的苯基選擇性和改善生物堿峰型的特點,但流動相精度過高,可操作性不強,一致性較難控制,在麻黃含量測定的實際應(yīng)用中麻黃堿與偽麻黃堿的分離并不穩(wěn)定,沒有達到理想的分離效果。此外,過高的成本和過強專屬性也限制了應(yīng)用。相反C18反相色譜柱其理論塔板數(shù)可以達到10000以上,同時對稱性(拖尾因子)和分離度均符合藥典要求。
3.2 波長與柱溫
分別取鹽酸麻黃堿和鹽酸偽麻黃堿,經(jīng)紫外光譜掃描,結(jié)果顯示兩種對照品均為末端吸收。參考2015年版《藥典》將210 nm作為測定波長。實驗考察了不同溫度對色譜分離的影響(20 °C、25 °C、30 °C 和 35 °C),結(jié)果表明溫度變化不會造成兩生物堿色譜峰的變化,因此不作溫度限制。
3.3 含量
2005版藥典規(guī)定鹽酸麻黃堿含量不得低于1.0%,而2010版將色譜柱換成極性乙醚柱后,增加了鹽酸偽麻黃堿的含量測定,并規(guī)定兩個成分的總含量不得低于0.8%,即單一成分的含量限度高于兩個成分總含量限度,其中緣由仍然值得進一步研究商榷。
3.4 保留時間
采用該色譜方法雖然準(zhǔn)確、穩(wěn)定且簡便易行,但仍然存在保留時間過長的不足,有待進一步完善。
