
計算模擬在鋰金屬負極研究中的應用
2021-03-08 01:42:34華廣斌樊晏辰張千帆
物理化學學報 2021年2期
關鍵詞:界面
華廣斌,樊晏辰,張千帆,*
1北京航空航天大學材料科學與工程學院,北京 100191
2南方科技大學微電子學院,廣東 深圳 518055
1 引言
鋰離子電池憑借自身容量大,工作電壓高,以及循環壽命長等優點,成為最常用的電化學儲能設備之一。便攜式電子產品和電動汽車市場的繁榮,為鋰離子電池提供更多的商業需求。與此同時,高能量密度、安全可靠等要求日益凸顯。在過去二十年,傳統鋰離子電池的石墨負極已經接近其理論容量(372 mAh·g?1),并且首次充放電時容量損失很大且不可逆。石墨負極已難以滿足新時期下的應用需求,而鋰金屬負極在該方面具有極大優勢:低電化學勢(?3.04 V,相對于標準氫電極)、低密度(0.51 g·cm?3)和十倍于石墨的理論比容量(3860 mAh·g?1),從眾多材料中脫穎而出,以鋰金屬作為負極的鋰金屬電池(lithium metal batteries,LMBs)引起許多科研工作者的關注。然而,鋰金屬電極的應用一直面臨諸多挑戰,例如,充放電循環中體積膨脹,鋰金屬與電解液之間界面反應復雜,電沉積過程中負極表面枝狀晶體生長難以控制,甚至存在“死鋰”(圖1a)1-4等。這些都嚴重影響了鋰金屬負極的安全穩定和高效。目前,科研工作者已提出許多方法解決上問題:調控固體電解質膜(solid electrolyte interphase,SEI)的成分和結構5-9、改進電解質的性質10-14、構建三維親鋰框架5,15-19等等(圖1b)。但是,由于鋰金屬負極的相關反應機理尚不完全清楚,如與電解液的相互作用、鋰的沉積等,許多關鍵問題無法從根本上解決。
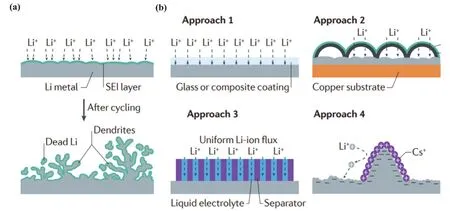
圖1 (a)鋰負極失效機制;(b)抑制鋰枝晶生長并改善界面穩定的方法:使用玻璃或復合材料作為表面涂層,使用碳或石墨烯薄層作為表面涂層,鋰離子傳輸均勻化,使用Cs+作為電解液添加劑,整合3D組件或使用鋰金屬粉末1Fig. 1 (a) Failure mechanism of Li-metal anodes; (b) approaches to minimize Li-dendrite growth and improve the interfacial stability: surface coating with glass or composite, surface coating with thin carbon or graphene layers,uniform Li-ion flux, adding Cs+ to the electrolyte, incorporating 3D patterns or using Li-metal powder 1.
計算模擬,特別是密度泛函方法、分子動力學方法,可以預測材料性能、模擬反應過程,成為篩選材料、揭示反應機理的有效工具之一。近年來,理論計算的應用愈加成熟,針對計算模擬的專業軟件在化學、材料、醫學等領域的應用愈加廣泛。許多工作將理論計算和實驗相結合,取得很好的結果。
將理論計算應用于鋰金屬負極的研究,有助于從根本上認識并解決上述關鍵問題。具體而言,研究的目標主要為:(1)鋰金屬與電解液的界面反應;(2)SEI和保護層的性能;(3)鋰離子在電極表面的成核沉積20。本文以此對近年計算模擬在鋰金屬負極中的應用進行綜述。
2 計算方法
Shi等21指出,基于理論算法、建模仿真以及計算機技術的進步,鋰離子電池領域中材料、電池、設備和電池組正通過大型數據共享逐步結合,這將帶來廣泛而深遠的變革,加速整個產業鏈的發展。在此背景下,多尺度建模仿真顯得尤為重要,有必要明確各尺度下主要的計算建模方法。原子尺度模擬上,一般涉及三種計算方法:第一性原理計算,分子動力學方法和蒙特卡洛方法;在中尺度上,可以使用相場模擬的方法,該方法可以實現從原子尺度到連續體尺度的更大范圍建模;對于宏觀尺度,通常研究電池系統中的應力應變及熱行為等,因此該尺度下主要使用連續介質理論,有限元方法和有限差分方法21。明確不同尺度的特點、理論基礎和計算方法,對針對性地選擇方法進行鋰電池計算模擬有指導意義。本篇綜述主要在原子層次討論計算模擬在鋰金屬負極中的應用,第一性原理計算和分子動力學方法是主要使用的計算方法。
基于密度泛函理論(Density function theory,DFT)的第一性原理計算可以精確計算固體系統的電子基態,因此在LMBs的計算模擬中應用廣泛22。1965年,Kohn和Sham23在Thomas24、Fermi等前人的研究基礎上,提出了著名的Kohn-Sham方程,揭示了基態能量和電子密度間的確切關系。通過自洽求解Kohn-Sham方程,可以獲得材料的總能量、穩定結構和能帶等基礎信息。特別地,對于LMBs,通過計算材料缺陷的形成和遷移能量,能夠預測相的穩定性;通過對相關化合物的生成焓進行計算,能夠預測熱力學上的穩定性;通過對不同晶格參數的能量變化和加載力進行計算,可以獲得材料的機械性能22,25。此外,還可以根據鋰不同狀態的能量差異對電壓進行預測26。
建立Kohn-Sham方程需要一些近似處理和定理支撐。首先,對于多粒子體系,在不考慮其他外場作用的前提下,定態薛定諤方程中哈密頓算符為



根據Born-Oppenheimer近似28,將原子核的運動和核外電子的運動分開考慮:研究電子運動時原子核處于其瞬時位置上;研究原子核運動時不考慮電子在空間的具體分布。因此,對于電子,有
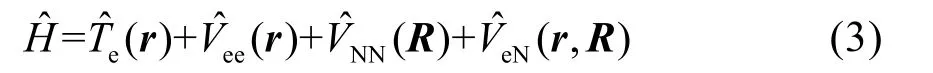
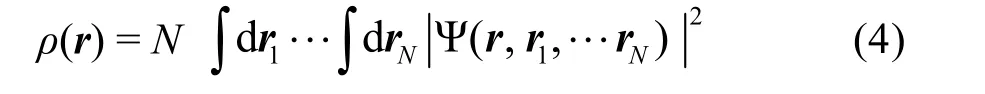
由此,體系的能量可寫作

其中,假定所有電子都具有相同的局域勢v(r) (包括電子之間的關聯、原子核勢場、外場等),使用表示外勢對電子的作用,有

使用無相互作用電子系統的相關量替代上述系統,并用N個單電子波函數φi(r)組成密度函數ρ(r),即故電子動能

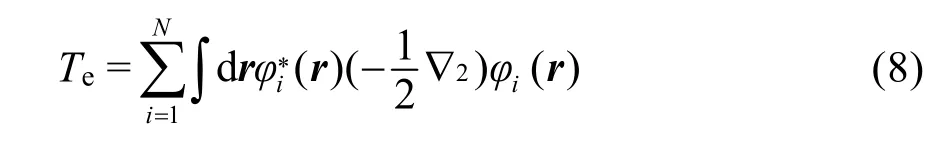
電子間相互作用

用交互關聯項Vxc表示其余相互作用及替代過程中損失的復雜性。由變分法,得著名的Kohn-Sham方程23
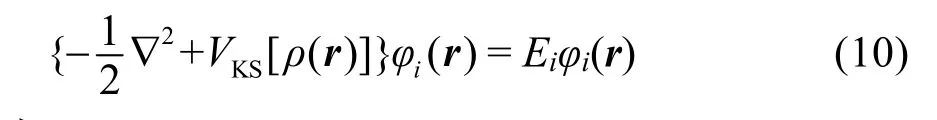
其中
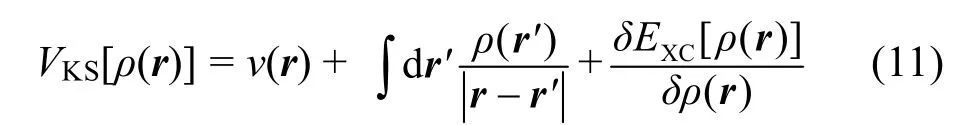
求解Kohn-Sham方程的難度主要在于,其中含有交換關聯勢能VXC,目前尚不知道確切形式,但已有許多有效近似方法。例如,局部密度近似(LDA)假設VXC只與局域密度有關而與整個體系密度無關,通過求解均勻電子氣來構造VXC的解析形式。因此LDA一般試用于電子密度變化比較平緩的體系,能夠有效處理金屬,但對多數氧化物和鹽存在較大偏差。Perdew28提出了廣義梯度近似(GGA),在VXC中引入電子密度梯度,以此來改善誤差。每個小范圍的VXC不僅僅依賴于其自身的局域電子密度,同時也受到周圍電子密度的影響。這使得近似更符合真實情況。GGA目前已衍生出多種形式,例如PW8629、PW9130。當系統中含有過渡元素或稀土元素時,系統的d,f軌道電子會趨于局部化,電子間的相互作用會使傳統方法產生較大偏差,此時DFT + U方法更為有效,其中U是庫倫作用參數,一般通過帶隙等實驗數據擬合得到。當LMBs中含有過渡元素或稀土元素時,可以嘗試使用該方法31。改善LDA、GGA兩種近似的另一種方式為考慮雜化泛函,將GGA的交換能EXGGA與非局域的Hartree-Fock型交換能EXHF以一定比例α混合,和GGA的關聯能EXGGA,共同構成交換關聯能EXC,如下所示
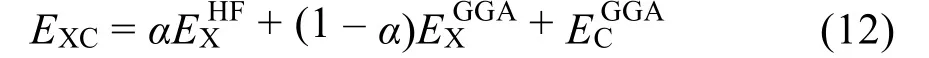
進一步將EXHF限制在短程內,即為HSE型雜化泛函32,在計算帶隙方面更加準確,被廣泛使用。但雜化泛函在處理性質相差較大的異質結構上并不理想,GW近似可以解決這一問題。實際中,GW近似在DFT計算后作為微擾執行,由此得到的能帶計算結果與實際相符得很好。
以上為DFT的主要理論和方法,在實際應用中,許多計算程序已被封裝,方便調用,例如VASP(ViennaAb-initioSimulation Package)是目前進行DFT計算較為完備、最流行的商用軟件之一,用戶只需向其中輸入研究對象、計算方法和待計算性質等命令即可進行計算。但合理應用的關鍵在于正確設置參數、選擇合適的求解方法。例如,Shi等33將DFT與應力輔助擴散方程相結合,對幾種集電器和相關合金中的鋰擴散機理進行研究。其中根據金屬材料(銅、銀、鋅等)特點、研究目標(體積變化、鋰傳輸路徑等),使用GGA近似方法,并確定收斂判據、k點設置等相關參數。
分子動力學(molecular dynamics,MD)方法重點在于對短時間內微觀動力學行為的模擬,是研究復雜系統的統計性質和擴散行為的有效工具之一。MD方法根據系統自身的動力學規律,模擬粒子間的相互作用和運動狀態,對于多數材料來說,可以將電子運動與原子核運動分開考慮,前者使用量子力學方法處理,后者使用經典力學的方法處理;最后根據統計物理學規律,建立粒子的位置和運動狀態等微觀量和溫度、比熱容等宏觀量的關系。因此,MD方法在處理粒子擴散行為中的路徑、能壘等關鍵問題方面十分有效。具體地,可以利用MD模擬研究LMBs中鋰的擴散、電極與電解質的界面反應等現象34-36。該方面相關工作在后文詳細綜述。除MD以外,微動彈性帶(nudged elastic band,NEB)理論也常用于計算模擬,用于研究鋰的擴散行為37。同MD相比,NEB可以更為有效地模擬高活化能的反應38,但不能處理復雜的、含有太多粒子的系統。
過渡態理論和MD方法易獲得分子間反應過程和分子擴散路徑、能壘等動力學信息,但結果的準確性很大程度受選用的勢函數影響。對于復雜的鋰化界面,勢函數的確定更為繁瑣,可直接使用的結論較少。此外,需要對適用于研究體系的模擬步長進行測定。這些都限制了過渡態理論和分子動力學方法的應用范圍和效率。
這里以Xiong等39對Cu(111)表面性質和鋰離子遷移進行計算模擬為例,說明DFT和MD應用中的相關細節。該工作以VASP為計算工具,首先使用VASP計算塊狀Cu的結構參數,將結果和文獻相比較,驗證計算方法的可靠性和可移植性。根據體系特點,所有計算均基于PW91-GGA進行,并設定截斷能(即考慮波函數展開結果的有限項數)、k點選取方式和收斂判據等計算參數。在研究擴散之前,Cu的表面能和功函數等特征進行計算,并判斷收斂性,進一步保證計算的有效性;之后移除部分銅原子,模擬空位的狀態。在此基礎上,利用MD方法對鋰離子運動進行動態模擬,以準確獲取擴散能壘,最后根據模擬結果進一步研究擴散行為,如最佳路徑等。該工作在計算模擬的應用中具有代表性。
與材料基因工程關系密切的高通量計算方法是材料計算模擬的全新模式,可以在大范圍內篩選樣品,降低實驗成本,縮短材料研發周期。其關鍵在于通過預先計算一系列樣本,提取其中的屬性或與屬性相關的信息40。通過將高通量計算與機器學習結合,可以減少不必要的計算,進一步提高篩選效率41。Correa-Baena等42提出了未來材料研究方法的愿景:首先,自動化實驗對候選材料進行高效測試;接著,高性能計算通過預測推斷材料相關屬性,根據有潛在價值的材料確定實驗范圍;最后,機器學習將兩者結合,用實驗結果自動完善理論并幫助確定下一個實驗。目前,已有許多科研工作者向這一方向邁進。例如,Severson等43應用機器學習工具針對電池的循環壽命進行預測和分類。該工作證明了將實驗數據獲取和數據驅動建模結合以預測復雜系統行為的可行性。在利用機器學習方法研究并預測優良固態電解質方面取得較多進展,將在本文的固態電解質部分進行綜述。
隨著理論的發展,計算模擬的方法在LMBs的機理研究、優化設計等方面應用日益廣泛,例如利用MD方法揭示電解質與電極的界面反應機理44-46、通過第一性原理計算指導設計穩定高效的鋰電極保護層47-54等等。在這些研究工作中,計算模擬與實驗觀測相輔相成,共同推進LMBs領域的研究。一方面,計算模擬有助于揭示現有觀測技術難以表征的微觀機理,以及模擬難以實際操作的特殊實驗,而且通過計算模擬的預測和篩選,可以大大減少研究的時間和費用;另一方面,實驗為計算模擬得到的結論進行論證,并且實驗取得的結果有助于建立新的模型、開發新的方法。未來,計算模擬在該領域將會有更為廣泛的應用。
3 復雜的界面反應
由于鋰金屬的高反應活性,常規有機電解液會在鋰金屬電極上自發分解,同時伴有各種可燃氣體(如烷烴、烯烴、一氧化碳等)生成逸出,導致電池容量下降,并存在安全隱患。認識電解液和鋰金屬電極間的界面反應,對于設計有效的SEI、構建穩定的鋰電極至關重要55。分子動力學(MD)方法可用于模擬特定反應過程、研究其界面特性,是處理這類問題的強大工具之一。
為建立穩定的界面,應首先認識電解液溶劑和鋰金屬電極間的界面反應機理。目前,被廣泛應用于LMBs電解液的主要為酯類溶劑和醚類溶劑兩大類,此外還有一些新開發出、不易燃的特殊溶劑56。
酯類溶劑,例如環狀的碳酸丙烯酯(PC)和碳酸乙烯酯(EC)以及鏈狀的碳酸二甲酯(DMC)、碳酸甲乙酯(EMC)等,由于其最高非空分子軌道(highest occupied molecular orbital,HOMO)能量較低,對正極有很好的氧化穩定性,在LMBs中應用廣泛56。1,3-二氧戊環(DOL)和1,2-二甲氧基乙烷(DME)是兩種常用的醚類電解質。Chen等44根據DOL/DME工作時的分解現象,提出并驗證DOL/DME先吸附鋰然后分解的反應機理,并且發現DOL因分解勢壘更低,更容易分解為乙烯。此外,實驗觀察到,當DME與DOL比例增加時,鋰硫電池的放電平穩期和電容量均有提升。Camacho-Forero等45研究了DME溶劑在富電子環境下的分解機理,對最相關的分解反應進行熱力學和動力學研究。利用MD方法發現,DME暴露在鋰原子環境中,與鹽碎片反應時,熱力學上更易分解。目前已被證實,高濃度的DME有利于鋰電極的穩定46。根據MD方法,在高濃度電解液中,大多數陰離子以配位形式存在,而所有的鋰離子都以溶劑化形式存在。這些工作有助于深入認識酯類溶劑在界面反應中表現出的性質。
醚類電解液溶劑電導率高但高壓(4 V)下易分解,酯類電解液溶劑穩定性相對更好但電導率相對較低,需要將兩者的優勢相結合,設計高電導率、高穩定性的溶劑。之前的研究工作主要通過將多種化合物直接混合,但結果并不理想57。為此,Amanchukwu等58合成出由醚段和氟段組成、兼具高電導率和高穩定性的氟化醚類電解液溶劑(圖2a,b)。通過設計醚段、氟段的長度和構型并將兩者用共價鍵鏈接,以此研究新型溶劑的分子結構與電導率、穩定性的關系。該類電解質實現了在高達5.6 V的氧化穩定性下,2.7 × 10?4S·cm?1(30 °C)的高電導率。MD方法進一步揭示電解液中相關反應的細節,如離子的溶劑化環境等。根據鋰離子在不同電解液中的徑向分布函數(RDF) (圖2c-f)可以發現四甘醇二甲醚和氟化醚中醚鍵與鋰離子容易相結合,證實了前期提出的氟化醚中醚鍵與鋰離子配位并促進其傳輸的假設。此外,由于氟的強烈吸電子效應,醚鍵中氧的電子濃度被降低,不易被氧化,從而在整體上提高了該溶劑的氧化穩定性,這一點已在實驗上得到證實。該工作具有創新性,為設計新型電解液溶劑提供了新的思路。
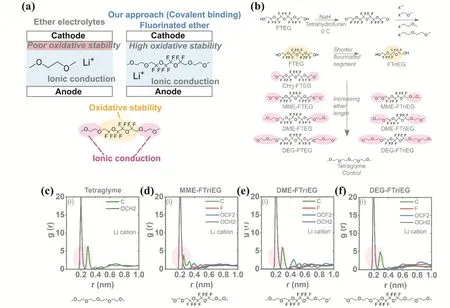
圖2 新型溶劑氟化醚的(a)設計思路;(b)合成方法;不同電解質中0.1 mol·L?1 LiFSA的徑向分布函數(RDF):(c)四甘醇二甲醚、(d) MME-FTriEG、(e) DME-FTriEG和(f) DEG-FTriEG 58Fig. 2 (a) Design idea and (b) synthesis method of new solvent fluorinated ether. Radial distribution function (RDF) of 0.1 mol·L?1 LiFSA in different electrolytes: (c) tetraglyme, (d) MME-FTriEG, (e) DME-FTriEG and (f) DEG-FTriEG 58.
電解質添加劑在界面反應和SEI的形成中起著重要作用。Camacho-Forero等59-62首先研究了在碳酸亞乙烯酯(VC)等添加劑存在下鋰電極與電解質的界面反應,該方法在穩定界面上取得一定成功,實驗中VC在鋰金屬表面聚集形成穩定堅固的保護膜,其作用在后續得到進一步研究和驗證。例如,乙腈(AN)由于具有很好的負極穩定性和離子電導率,常被用作電解液,但即便是在高濃度的情況下,仍對鋰金屬電極不穩定63;為此Peng等63向高濃度AN電解液中加入添加劑VC,利用MD方法進行研究。計算模擬證明了電解液中的鋰鹽與AN形成緊密配合的溶劑化結構,在初始充放電過程中與鋰金屬電極發生界面反應,形成致密穩定的聚碳酸酯類SEI,大大抑制了鋰的消耗。實驗中電池在0.2 mA·cm?2的電流密度下實現高達99.2%的庫倫效率,由此設計的Li??NMC622也具有很好的循環性能。
氟元素的引入可以改善鋰金屬電極的工作性能。盡管氟的電負性會降低電解液溶質的最低空分子軌道(lowest unoccupied molecular orbit,LUMO)能量,使溶質的還原穩定性降低,但是,氟可以以LiF的形式參與形成SEI,而實驗表明,LiF對SEI的穩定性、電池的循環性能等有著復雜的影響56。氟代碳酸乙烯酯(FEC)已被證實在形成富含LiF的SEI、穩定鋰電極方面表現出色。Zhang等10對富含LiF表層的形成過程進行動力學研究(圖3a-d),發現相比于電解液EC/DEC,添加劑FEC具有更低能量的LUMO,能更好地在鋰電極上分解,促進形成致密表層,利于鋰的均勻沉積(圖3e),從而使鋰金屬電池在長循環下依然高效穩定工作。
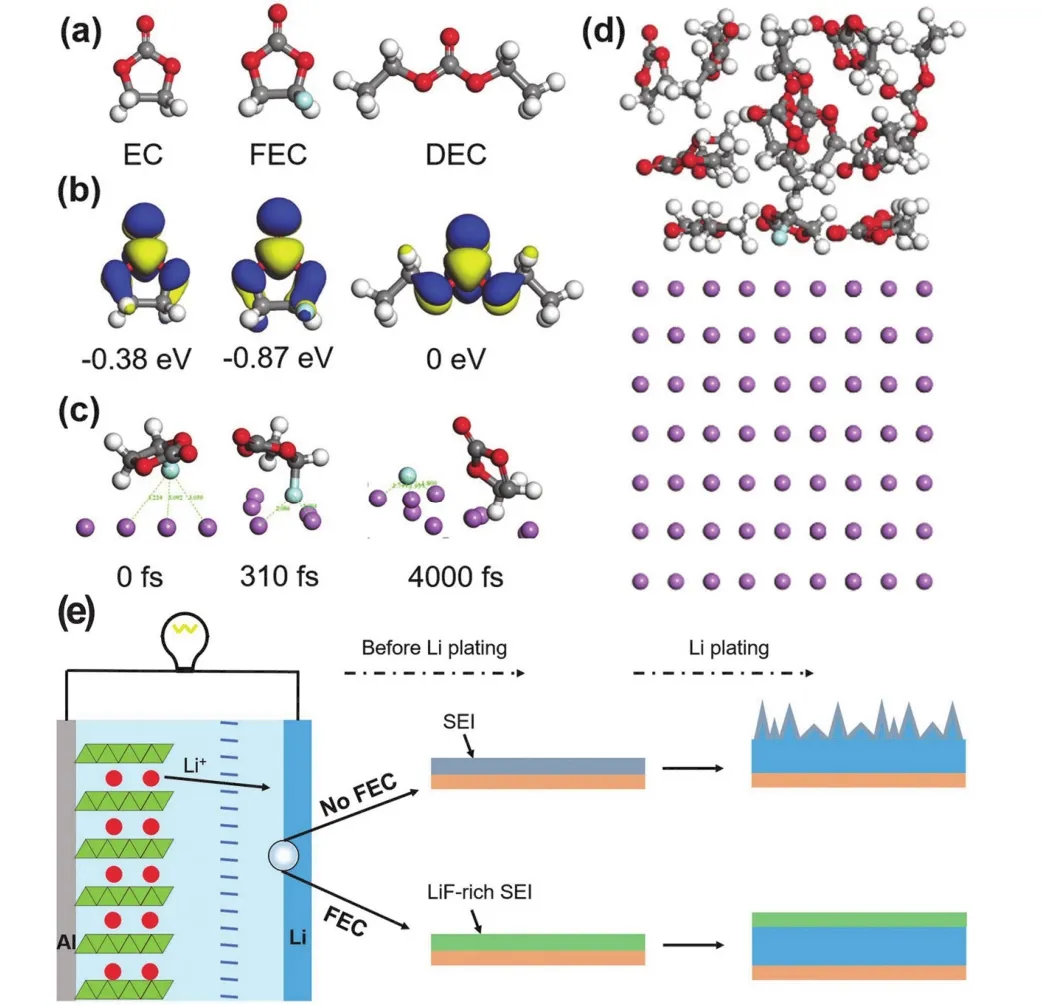
圖3 (a) EC,FEC和DEC的分子結構;(b) LUMO和EC,FEC和DEC的相對能量;(c)從頭算分子動力學模型;(d)在鋰陽極上分解的FEC分子的完整序列;(e) FEC添加劑對鋰金屬陽極的影響10Fig. 3 (a) Molecular structures of EC, FEC, and DEC; (b) visual LUMO and corresponding relative energy of EC,FEC, and DEC; (c) ab initio molecular dynamics model; (d) complete sequence of FEC molecule decomposed on Li anode;(e) schematic illustration of the effect of FEC additives on Li metal anode 10.
近日,Kim等64向富鎳正極的電池體系中引入3-(三甲基硅烷基)-2-惡唑烷酮(TMS-ON)添加劑,用以促進LiPF6(因其很好的離子電導率和氧化穩定性等優點而作為一種常見的商用電解質65)解離,抑制參與配對的LiPF6水解(產生HF等酸性化合物,影響電池性能)并清除電解液中的HF。實驗觀測和理論計算得到的結果符合預期。他們進一步研究了結果背后的機理。一方面,利用DFT計算,分析了電解液中TMS-ON對LiPF6的作用機理。通過計算電解液中各組分與鋰離子相互作用的形成能,確定各組分的熱力學最穩定構型,發現TMS-ON-Li+配合物具有最低的能量(?0.81 eV),最為有利,并進一步利用DFT計算各組分(EC、EMC、DEC、VC、ON和TMS-ON)對PF5的穩定作用(圖4a);另一方面,為了確認PF5對抑制LiPF6水解的作用,計算了有無TMS-ON-Li+配合物存在時PF5與H2O反應的能量變化(圖4b),從熱力學上證實了判斷。
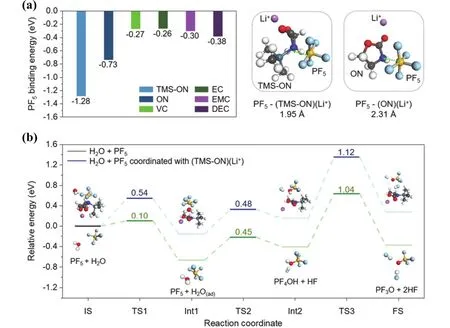
圖4 (a)與TMS-ON、ON、VC、EC、EMC和DEC同Li+配合時,PF5熱力學最穩定狀態的結合能;(b) PF5水解的反應能量64Fig. 4 (a) Binding energies of the most thermodynamically stable configurations of PF5 with TMS-ON, ON, VC, EC, EMC,and DEC coordinated with Li+; (b) reaction energy diagram for the hydrolysis of PF5 with and without TMS-ON-Li+ 64.
鋰鹽的種類和濃度也會影響電解液的分解以及電極表面保護膜的形成。Camacho-Forero等66對Li-DME/LiTFSI和Li-DOL/LiTFSI兩種界面反應分別進行研究,計算模擬的結果表明,LiTFSI能夠立即與鋰金屬電極反應,迅速分解生成LiF保護膜。同時,實驗沒有觀察到DME和DOL進行任何分解反應。此外,電解質自身結構也會受LiTFSI濃度影響67:低濃度時主要為反式結構,而在高濃度時主要為順式結構。這種順-反結構的變化影響了溶劑體系,進而影響鋰金屬表面上的界面反應。Zhang等68向電解質中引入FEC和硝酸鋰(LiNO3)以調節鋰離子溶劑化殼層中的成分以及FEI?的溶劑化結構,從而促進FEI?完全分解,形成穩定均勻、富含LiF和LiNxOy的SEI;并且根據MD方法和第一性原理計算,驗證了溶劑化殼層中的成分和結構。實驗中,由此設計的電池在庫倫效率、循環壽命方面表現優異,循環的穩定性大幅提高。Chen等69通過第一性原理計算并結合原位光學顯微鏡觀測,發現鋰離子-溶劑配合物的存在會極大促進電解質分解和氣體逸出。研究表明該機制也同樣適用于其他離子溶劑系統,包括鋰或鈉金屬電極與PC、DOL、DME或TEGDME電解質。這些工作加深了人們對LMBs中電解質的認識。
除電解質組成、濃度以外,電池的工作參數也會影響界面反應的動力學特征。溫度一直是影響界面反應的重要因素,而且對于在偏離室溫下工作的鋰電池尤為重要。Wang等70對高低溫鋰電池的運行性能進行研究,發現醚基電解液的鋰電池在60 °C下,平均庫倫效率為99.3%,可實現三百多個穩定循環;而在20 °C下,庫倫效率在75個循環內急劇下降。電子顯微鏡觀測顯示,在60 °C時SEI出現了特殊的納米結構,能夠抑制工作過程中的副反應,保證循環穩定并維持低電化學阻抗。之后,Gao等71將1,3-苯二磺酰氟自組裝單分子膜結合到銅基底上(EAM Cu),形成多層SEI,有效密封了鋰金屬電極表面,實現?60到45 °C溫度范圍內鋰均勻沉積,保證電池的電化學穩定。利用MD方法研究低溫狀態下的界面反應發現,EAM Cu較裸銅反應能變化更小,從而有效抑制電解液分解。另一方面,通過定量核磁共振和MD模擬監測各電解質組分的消耗速率,發現EAM Cu可以降低電解液消耗,因此提高了鋰電池的穩定性。該工作為低溫充電電池的發展開辟了一條新的道路。此外,Wang等72報告了LiTFSI/DOL-DME電解液中鋰金屬負極的表觀循環性能,并通過微電極方法(microelectrode technique)獲得相關動力學參數。研究發現合適的動力學參數(如遷移數、電流密度、成核速率和腐蝕速率等)及其良好的匹配度有利于提高鋰金屬負極的循環性能。該工作揭示了鋰電極在電沉積過程中基本參數和表觀循環性能間的關系,為建立新模型和計算模擬提供了新見解。
為防止鋰金屬的過量消耗,鋰硫電池中的鋰電極還可用聚硫化物作為表層調控界面反應73,74。Camacho-Forero等66對電解質中的聚硫化物與鋰電極之間的界面反應進行深入研究,發現線形或環形的Li2S8會在表面被完全還原為Li2S。之后,Liu等75從理論上對Li2S/Li界面進行計算模擬,闡明了在電池工作過程中由于長鏈聚硫化物的分解,Li2S會在鋰電極表面形核、生長成膜。同時,他們發現Li2S在Li(110)表面更易形成無序夾層,而在Li(111)表面則更易生長為完美的Li2S(111)。
4 固體電解質膜
由于鋰金屬獨特的活潑性,大多數有機溶劑會與之反應,在初始充電/放電過程中形成可傳輸離子但不能傳輸電子的固體電解質膜(solid electrolyte interphase,SEI)76。構建穩定的SEI是抑制鋰枝晶生長的有效途徑之一,因此了解SEI的形成機理、組成和穩定性十分重要74,77,78。對于實際中遇到的諸多挑戰性問題,理論模擬是人們深入認識機理、尋找解決方案的有效工具。具體地,可以基于第一性原理計算,深入認識SEI的表面化學反應、表面形態、電化學性質以及動力學特性等等。
對于SEI,鋰離子的傳輸機制是最基本的問題之一,這需要深入了解SEI的結構特點。鋰金屬負極表面的SEI主要為電解液中有機物(及添加劑)與活潑的鋰金屬在初始充放電過程中反應生成,通常由相對致密的無機層(含Li2CO3、LiF等)和相對疏松的有機層(含ROLi等)兩部分組成。Shi等79為此開發了一種多尺度理論方法:首先根據相關的實驗表征結果建立雙層/雙機制模型,進而在中尺度上建立擴散方程,并結合實驗結果80加以驗證。基于上述方法,科研工作者進一步研究了鋰離子的傳輸機制和電子的傳導特性,取得重要成果:(1)在一定電壓范圍(0-4.4 V)內,Li2CO3中鋰離子的主要擴散方式由電壓決定。盡管陰極和陽極的SEI成分相似,但主要的鋰離子擴散載流子卻不同:在0.98 V之下以間隙擴散為主,而在3.98 V之上以空位擴散為主81。(2)在雙層結構的SEI中,存在兩種不同的鋰離子傳輸機制:多孔的外層可以傳輸鋰離子和陰離子,而致密的內層只能傳輸鋰離子82。(3)通過計算電子自鋰金屬到SEI中各種絕緣組分的電子隧穿勢壘,發現SEI的形成和持續增長都會導致電池循環工作過程中容量的不可逆減少83。這些工作有助于人們深入認識SEI的形成、結構和性質。之后,Zhou等84利用液態二次離子質譜和MD方法計算模擬,確定了SEI形成過程中的動力學圖像。在初始充電過程中,溶劑分子進行自組裝,并在鋰離子和電極表面電勢的誘導下,在電極/電解質界面形成雙電層。雙電層結構進而影響了最終SEI的結構和性質:呈負電性的電極表面從內層排斥陰離子,從而促進形成致密、超薄的無機內層,也正是該層決定了SEI離子導通、電子絕緣的性質。之后進一步形成了相對疏松的有機外層。此外,在高濃度、含氟電解液中,由于雙電子層中存在陰離子,無機內層具有高濃度的LiF。該工作深入揭示了SEI的形成機理,并且該觀測方法可以在納米尺度上實時觀測界面層,為進一步研究鋰電池SEI提供了有效的觀測手段。
對于一些常見SEI,它們的基本性質,如機械強度、鋰擴散率和電子結構等85,已被人們掌握,但當這些材料應用于鋰金屬表面時,復雜的界面效應可能會使上述性質發生很大變化。Liu等49基于DFT計算,對鋰金屬與LiF和Li2CO3之間的界面進行研究,實驗結果表明,LiF/Li界面自鋰電極到SEI的電子隧穿能壘更高,而Li2CO3/Li界面的機械強度更高。Li2CO3和LiF之間的接觸促使電荷沿界面累積,進而產生高濃度的離子載流子,顯著改善鋰離子傳輸效能、減少電子泄漏50。
除上述界面效應外,科研工作者從尺寸效應、電壓效應和界面極化等角度深入研究。Pan等51利用空間電荷效應設計由LiF和Li2CO3組成的人工SEI。他們發現,可以通過減小SEI中Li2CO3晶粒的尺寸,改善離子載流子,進而將離子電導率提高數個數量級。Leung等48研究了電壓對雙電層的影響,進而提出通過控制表面偶極子來改善電極鈍化的策略。Jand等53研究了在石墨烯上生成的LiF的結構和機理,發現SEI的原子結構受靜電作用控制,并且可以通過簡單的靜電模型預測SEI的穩定性。Zhukovskii等52模擬了在二維極性界面上引入多余鋰原子的情況,對界面進行了DFT-LCAO比較計算。該模型也證實了電池在低電位時具有額外的存儲容量。此外,Simeone等54首次設計出合理的雙層SEI。他們的工作基于實驗和理論,有助于在原子水平上認識金-電解質界面反應。
目前,人工SEI被廣泛應用于減弱界面反應、抑制鋰枝晶生長以及保護鋰電極。一方面,人工SEI可以通過先進的薄膜制備工藝覆蓋或包裹鋰金屬負極,避免鋰金屬與易反應的電解質直接接觸,在減少電解質消耗的同時提高金屬電極的化學穩定性;另一方面,人工SEI是鋰離子傳輸的必然途徑,可以對沉積過程加以控制,促進均勻沉積,并且可以通過高機械強度抑制枝晶生長,提高金屬電極的物理穩定性。理想的人工SEI應該滿足如下要求:(1)對于電解質中的絕大多數成分穩定;(2)能使鋰離子快速擴散,電導性能優秀;(3)具有足夠抑制鋰枝晶生長的機械強度,以及足以承受鋰金屬沉積的剛度。針對上述要求,有必要進行全面系統的理論研究以篩選出合適的材料用于設計人工SEI。特別地,由于人工SEI和鋰金屬接觸而導致的鄰近效應不能忽視。
之前的研究表明,LiF對SEI層的穩定性、電池的循環性能等有著復雜的影響,引起人們注意,但具體機理仍不清楚56。Ren等86系統研究了鹵化鋰(LiHa = LiF、LiCl或LiBr)參與構成的SEI的相關性質。利用第一性原理計算,分別計算模擬了均勻和非均勻兩種表面形態的LiHa-SEI的鋰沉積過程,研究了鋰的表面吸附、表面擴散以及電荷轉移等相關細節。研究發現,無論LiF-SEI的表面是否均勻,鋰均具有很高的遷移能壘,表現出很好的穩定性。該工作還在計算上證明由于LiF-SEI上的鄰近區域的變化對離子轉移能壘影響較小,LiF相比于LiCl或LiBr更適合作為SEI。Zhu等87利用第一性原理,全面地研究了由共價石墨材料(石墨烯和h-BN)和無機鹽(LiF、Li2O、Li3N和Li2CO3)組成的人工雙層SEI(BL-SEI),對關鍵的界面性質,如化學穩定性、離子電導率和機械強度,進行了系統研究(圖5a)。在所有考慮的BL-SEI中,石墨烯/LiF組合在計算上表現出最佳的界面穩定性和電化學性能。同時,計算模擬發現BL-SEI對鋰金屬負極的保護作用是各向異性和結構缺陷共同作用的結果(圖5b-e)。該工作在原子電子層面揭示了BL-SEI對鋰金屬負極的保護機制,為設計合理的人工BLSEI提供了指導。

圖5 (a) BL-SEIs;(b)石墨烯/LiF<111>的晶體結構;(c)石墨烯/LiF<111>的電子密度分布;(d)具有單個C缺陷的石墨烯/LiF<111>的結構俯視圖;(e)石墨烯/LiF<111>中石墨烯表面的電子密度分布87Fig. 5 (a) BL-SEIs; (b) atomic conformations of graphene/LiF<111>; (c) the differential charge density of graphene/LiF<111>; (d) graphene/LiF<111> with single C defect in graphene (top view); (e) plane electron density difference along the graphene surface of graphene(CD)/LiF<111> 87.
第VI主族元素的S也被應用于設計保護層,參與SEI形成66,73-75。Chen等88通過硫單質在鋰金屬電極表面形成Li2S保護層,兼具高離子導電率和均勻性。通過COMSOL仿真模擬發現,形成的SEI離子電導率越高鋰離子沉積越均勻,越有利于抑制鋰枝晶的生成。之后,Liu等89通過硫化硒與鋰金屬的氣固反應制得Li2S/Li2Se保護層,由DFT計算證明,Li2Se較Li2S的鋰離子遷移能壘更低,從而表現出更好的離子導電率。此外,在實際實驗中,具有Li2S/Li2Se保護層的LiFePO4、S/C及LiNi0.6Co0.2Mn0.2O2全電池在電池循環性能上均有提升。
除上述通過控制鋰化物設計人工SEI的方式以外,可以增添保護層進一步改進人工SEI的性能,例如前文Zhu等87在BL-SEI方面的研究工作。Tian等47基于第一性原理計算,研究了多種不同拓撲結構的層狀材料(石墨烯、h-BN等)構成的人工SEI。他們發現缺陷類型、晶體結構、環的尺寸以及接觸的金屬表面都會對人工SEI的諸多性質產生影響,例如空位影響h-BN層電子密度分布(圖6a-c),進而影響穩定性、鋰離子擴散能力和機械強度等。特別地,引入缺陷可以降低鋰離子的傳輸阻礙,促進鋰離子通過SEI擴散,但同時也會大大降低機械強度、剛度以及臨界應變和應力(圖6d,e)。缺陷的存在會顯著削弱h-BN在雙軸拉伸的等效載荷下的機械強度,而金屬的存在會大大降低鋰離子的擴散勢壘。進一步的分析證實,人工SEI中的電子和電荷分布會影響擴散勢壘。其中,鋰離子對SEI電子的捕獲能力在擴散勢壘中起決定作用。缺陷周圍的電荷密度越低,鋰離子擴散的阻礙越小。同時,鋰金屬中的電子可以轉移到保護膜上,填入反成鍵軌道,削弱人工SEI中的共價鍵,這是金屬鄰近效應的根本原因。這些研究為鋰離子與人工SEI材料之間的相互作用機理提供了原子尺度上的新認識47。
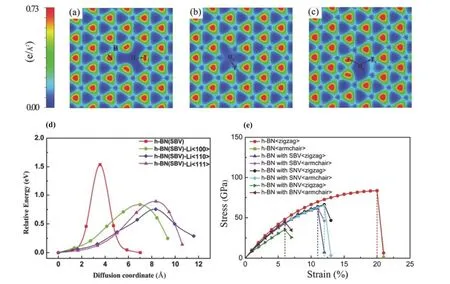
圖6 (a)含單個B空位(SBV)的h-BN電子密度分布;(b)含單個N空位(SNV)的h-BN電子密度分布;(c)含雙B-N空位(BNV)的h-BN電子密度分布;(d)Li+通過有/無鋰化的h-BN(SBV)比較;(e)有/無缺陷的h-BN應力-應變關系44Fig. 6 Charge density plots of (a) h-BN with SBV, (b) h-BN with SNV, (c) h-BN with BNV; (d) comparisons on Li+ ion diffusion through lithiated and unlithiated materials of h-BN with SBV; (e) the strain-stress relations for h-BN and defective h-BN 44.
對于液態電解質電池,隔膜是其重要組成部分,用于物理分隔正負極,避免兩者接觸,同時隔膜應允許離子自由傳輸,并能阻隔電子。隔膜上的表面涂層具有抑制鋰枝晶生長的作用,一些具有微納結構的改性膜因機械性能較好,能夠抑制枝晶生長90。鋰硫電池在實際應用中常存在硫電導率差以及由聚硫化鋰的穿梭效應導致容量降低等問題。通過對隔膜改性,可以調控聚硫化物的氧化還原,改善鋰硫電池的電化學性能。Hu等91設計一種功能性的氨化聚丙烯腈納米纖維隔板(APANF),利用DFT計算優化模型結構和相關成分的幾何構型,實現抑制鋰枝晶生長的同時控制硫化物的遷移。一方面,隔板由于氨基的存在可以很好地促進鋰離子均勻沉積,并且誘導生成富含Li3N的SEI,實驗中鋰的沉積形態呈球形且具有98.8%的庫倫效率;另一方面,纖維隔板由于支鏈的吸附作用,阻擋硫化物的遷移,使硫陰極的容量保持不變。Lv等92通過化學合成了NiCo2O4并將其涂覆在聚丙烯隔膜上,形成應用于鋰硫電池的新型隔膜。DFT計算模擬表明,NiCo2O4的表面具有較低的鋰離子擴散能壘,并促進聚硫化物進行一系列氧化還原反應。實驗進一步證實該隔膜可以極大地避免Li2S在鋰金屬表面上的沉積,從而減少鋰金屬和電解質的消耗,并顯著提高鋰硫電池的穩定性。Lei等93通過在聚丙烯隔膜上直接涂覆一層被還原的氧化石墨烯(reduced graphene oxide,rGO)/木素硫酸鈉(sodium lignosulfonate,SL)復合材料涂層,有效抑制帶負電的多硫化物傳輸,而不阻礙鋰離子傳輸。DFT計算模擬和實際實驗揭示并證實了其中的相關機制。Moorthy等94使用SnS2改性隔膜以促進聚硫化物中間體的氧化還原反應。DFT計算模擬顯示,SnS2對聚硫化鋰表現出很強的親和力,并促進其快速反應;實驗也進一步證實了該電池的良好循環性能。以DFT為主的計算模擬在鋰硫電池的隔膜改性中可以很好地研究聚硫化物氧化還原反應和親和性,這些工作為隔膜改性提供了指導。
5 鋰離子成核沉積
鎳、鋅等金屬電池在大電流充電下常常出現枝晶沉積現象,而活潑的鋰金屬更為顯著。目前關于鋰枝晶成核生長的模型主要有電荷誘導模型、離子擴散模型、電沉積異相成核生長模型和相場模型等。
電荷誘導模型由Ding等95提出:在鋰離子沉積過程中,受尖端效應影響,電荷易在金屬電極表面的尖端處聚集,產生局部電場,促進更多的鋰沉積,從而形成枝晶。Ding等95在此基礎上,提出利用陽離子尖端聚集產生的屏蔽作用抑制枝晶生長。根據能斯特方程
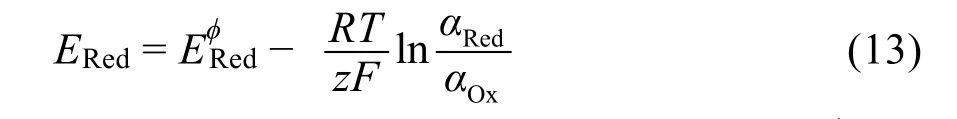
(其中ERed為被添加金屬陽離子的還原電勢,EφRed為其標準還原電勢,R為氣體常數,T為溶液溫度,z為轉移電子摩爾數,F為法拉第常數,αRed和αOx分別為還原態和氧化態的活度)可以通過降低被添加金屬陽離子的濃度獲得低于Li+的還原電勢,例如銫離子(Cs+)濃度為0.05 mol·L?1時,E(Cs+/Cs) =?3.103 V,低于濃度1 mol·L?1濃度下的E(Li+/Li) =?3.04 V。因此,當外加電壓在兩者之間時,被添加金屬陽離子吸附在較尖銳的鋰核上,促使鋰離子均勻沉積。Chazalviel等96,97從電解液中離子擴散的角度觸發,建立離子擴散模型:受電場等影響,LMBs兩個電極之間的電解質存在一定的陽離子濃度梯度;在達到臨界電流密度、耗盡鋰電極附近陽離子之前,電流只能持續很短的時間,之后電極表面的電中性被破壞,電荷在局部累積,從而形成枝晶。模型給出了耗盡Li+所用時間
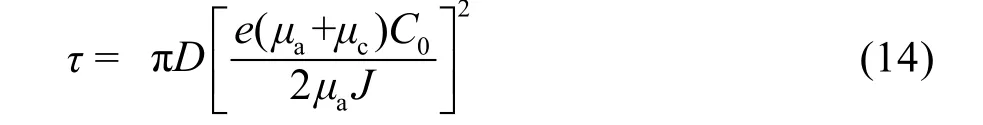
(其中D為擴散系數,μa和μc分別為陰陽離子遷移數,C0為鋰離子初始初始濃度,e為電荷量,J為電流密度)以及臨界電流密度

(其中L為兩極板間距),根據這一理論,可以用臨界電流密度J*預測枝晶生長。然而在現實中,即便電池通常在比J*小得多的電流中工作,枝晶也可能產生。這就表明,枝晶沉積是多種機制共同作用的結果98。Ely等99由基本原理建立了負極材料上電沉積異相成核生長模型,研究了成核和生長的條件和不同階段特征,并提出通過設計電極表面粗糙度和潤濕特性,以及控制電沉積電勢等方法抑制枝晶生成。Okajima等100建立了相場模型,對電極反應過程中電極電解質界面的動力學和表面形態進行研究,證實了超電勢是影響電沉積速率的因素,并發現電沉積的尖端半徑與生長速度的平方根成反比,該結論與凝固過程中的枝晶生長理論一致。這些模型有助于揭示電沉積過程中成核生長特征,為解決鋰電極的枝晶問題提供指導。
離子的擴散行為是研究鋰離子沉積形態的重要角度,還需關注鋰離子溶劑化結構、相互作用等具體細節。MD方法在處理粒子擴散行為中的路徑、能壘等問題方面十分有效,是研究鋰離子沉積的有效工具之一。目前已被證實,通過對脈沖電流進行優化,可大大延長LMBs的循環壽命。Qi等101利用分子模擬的方法,對這一過程涉及的機制做詳細研究。實驗證實,通過選擇合適的脈沖電流頻率和振幅,可以有效地控制鋰離子溶劑化結構,調控鋰離子的擴散,從而實現抑制枝晶生長。同時,他們證實鋰離子的擴散率受陰陽離子間的相互作用等多方面因素影響,而且這種相互作用有利于提高鋰離子的擴散率。此外,他們還發現工作電流的性質會極大影響電池的循環壽命,例如,將電池置于某些特定的脈沖電流波形下工作,可以使其循環壽命增加一倍以上。該研究加深了人們對電池在脈沖電流下工作機理的理解,為進一步研究鋰枝晶生長提供了新的視角。此外,Li等102提高電流密度(大于9 mA·cm?2),利用焦耳熱誘導鋰在電極表面遷移,平整表面枝晶,取得很好的效果。這也進一步說明枝晶沉積是多種因素復雜作用的結果,需要整體分析。Lin等103報道了鋰在異質導電表面上的快速腐蝕現象,并對相關機理進行研究,利用Kirkendall效應來解釋、描述腐蝕現象,同時實驗觀察到該機制也傾向于引起枝晶生長。
實際中,為金屬電極構建特殊的基體框架是調控鋰離子成核的動力學行為、抑制鋰枝晶生長的重要方法。DFT有助于揭示其機理,為合理設計金屬電極提供方向。
與Li-S和Li-O電池的正極類似,碳材料可以參與組成負金屬電極框架,誘導鋰成核,其中,石墨烯以其獨特的組織結構和導電性能,引起人們注意。但完整的石墨烯材料(PG)對鋰的親和力相對較弱,需要進行修飾、改造。Zhang等16將氮摻雜石墨烯(NG)作為基體材料進行研究。親鋰的含氮的官能團(如吡啶和吡咯氮)能夠誘導鋰成核,實現均勻沉積。實驗結果表明,使用NG的金屬負極,在循環過程中無枝晶產生并且具有較高的庫侖效率。Wang等104向石墨烯中引入硫原子,使PG轉變為硫摻雜石墨烯(SG),研究其上的鋰成核過程。通過第一性原理模擬發現,SG中摻雜的硫原子可以在大范圍內提高基體材料對鋰的吸附能力(圖7ac)。對于多個鋰原子在SG上的吸附行為,理論上硫原子可以在鋰的初始沉積過程中提供有五個成核位點,從而抑制鋰的不均勻沉積(圖7d-e)。實驗驗證了由此設計的鋰電極具有極低的超電勢(5.5 mV)和很高的面積容量(3 mAh·cm?2)。此外,Zhai等105合成出金屬摻雜石墨烯,將其作為基體材料,以調節鋰成核沉積,并利用DFT研究其中機理。引入到NG上的金屬原子不僅可以以適中的強度梯度增加金屬原子位點周圍局部區域的鋰吸附能力,而且還可以通過與N和C以特殊的配位形式提高整個材料的結構穩定性。Yi等106報道了一種通過激光處理石墨烯(LIG),調節鋰形核沉積動力學特性的方法。與普通的銅箔相比,LIG材料存在大量的缺陷和雜原子,大大降低了鋰的成核勢壘;同時,理論計算表明,在鋰的異相成核過程中,缺陷是成核中心。使用該材料的鋰電池具有極高的庫倫效率和穩定性。這一工作凸顯了成核動力學在鋰電極穩定性中的重要作用。
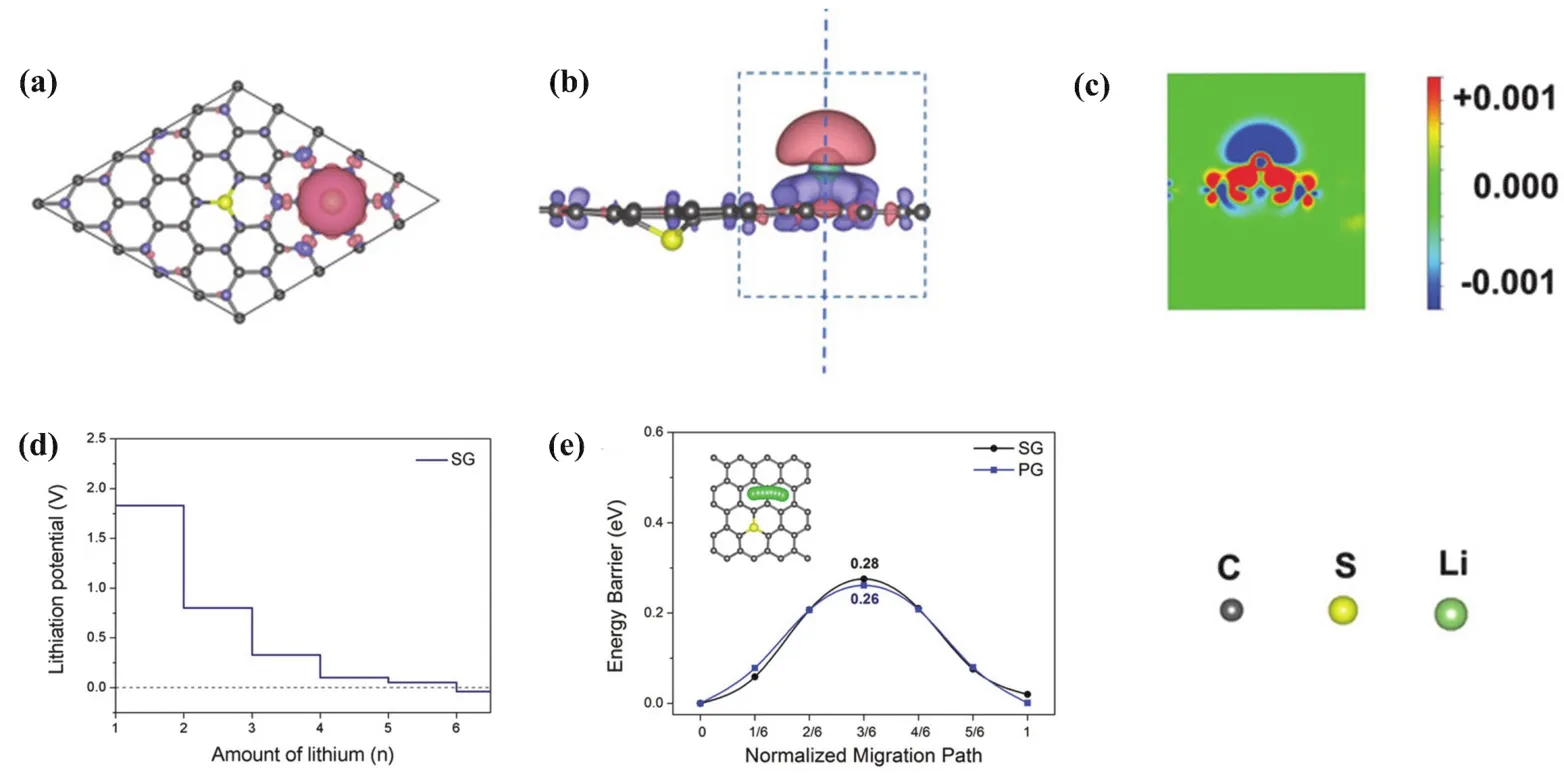
圖7 吸附了一個Li原子的SG的電荷密度差的(a)俯視圖和(b)側視圖,紫色和粉紅色區域代表電荷累積和損失區域;(c) (b)中藍色虛線處的平面上電荷密度差的截面輪廓,等值面值設置為0.001 e·??3;(d)鋰電勢隨SG上Li原子的數量而變化;(e)吸附的Li原子沿著SG路徑擴散的能量變化,插圖顯示SG上的Li擴散途徑104Fig. 7 (a) The top view and (b)the side view of the charge density differences of SG with one Li atom adsorbed,the violet and pink areas represent the charge accumulate and loss regions; (c) cross-section profile of the charge density difference at the plane across the blue dashed line in (b); the isosurface value is set to 0.001 e·??3; (d) the lithiation potentials changing with the number of Li adatoms at the SG; (e) the energy variations with the adsorbed Li atom diffusing along the pathway on SG, inset image indicates the Li diffusion pathway on S-doped graphene 104.
除二維石墨烯材料以外,碳纖維布也被用作鋰金屬負極的基體材料,在控制鋰均勻沉積方向效果顯著107。Ye等108向碳纖維布上引入RuO2金屬氧化物顆粒,以提高碳纖維布的親鋰能力。DFT計算結果表明,由于堿金屬與Ru的反應產物和鋰離子間存在較高結合能,可誘導鋰離子在碳纖維周圍均勻地沉積。Liu等109、Yue等110分別利用Co3O4、ZnO修飾碳纖維布,并結合理論計算和實驗證明其在提高基體親鋰性和促進鋰均勻沉積方面的作用。
多孔金屬基體材料在機械強度、結構穩定性以及電導率等方面具有先天優勢,近年來,在抑制鋰金屬體積膨脹、枝晶生長方面取得很好的成果111-116。商用的泡沫鎳材料因其比表面積較低和疏鋰的特性,難以投入實際應用,Ke等117通過電沉積鋰化的方式將親鋰的AuLi3顆粒均勻地覆蓋在在泡沫鎳骨架上,使基體材料表現出親鋰的特性。由DFT計算發現,AuLi3顯著降低鋰的成核能壘并增強了鋰沉積的均勻性,從而有效抑制鋰枝晶在泡沫鎳空隙內的生長。Lu等118在具有石墨碳氮化合物(g-C3H4)涂層的泡沫鎳(圖8d-e)上實現了鋰的均勻成核沉積。DFT計算和實驗結果表明,g-C3N4上的三均三嗪單元在空間形成微弱的負電場,對鋰離子有很強的捕獲能力(圖8a-c),大大降低了形核的超電勢(圖8f);g-C3N4的高含氮量使得材料表面存在大量均勻分布的鋰成核位點,從而誘導鋰的均勻沉積。基于g-C3N4@Ni泡沫的鋰金屬負極表現出極佳的電化學性能:電池在300次循環后庫倫效率仍達98%,壽命長達900 h。
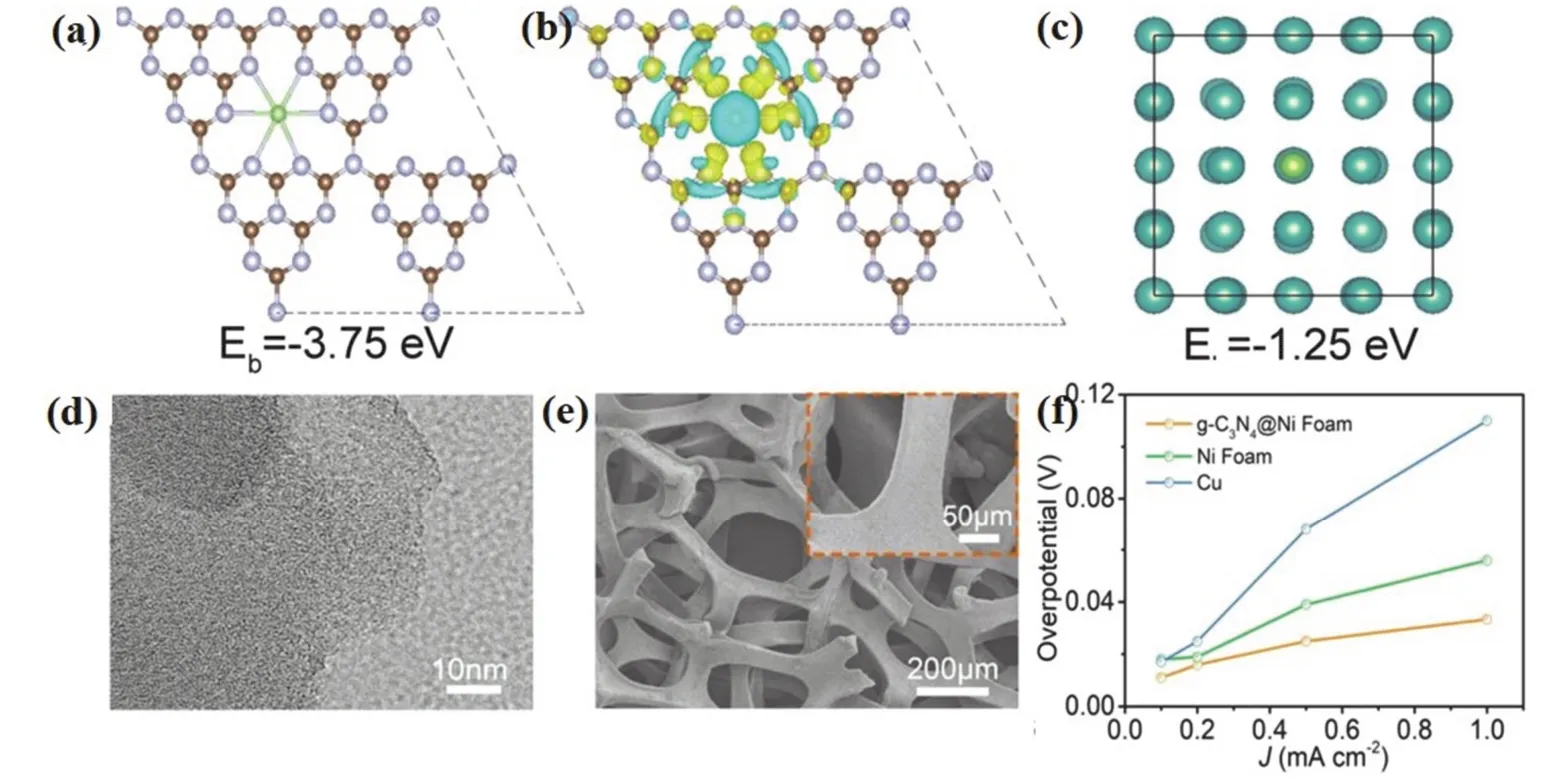
圖8 (a)用于計算吸附在g-C3N4上的Li +的結合能的晶體模型;(b)相應的電荷密度差(棕色、紫色和綠色的球分別代表碳原子、氮原子和鋰原子;黃色和淺藍色區域分別代表正電荷差和負電荷差);(c)用于計算吸附在Ni上的Li+的結合能的晶體模型(藍色和綠色的球分別代表鎳原子和鋰原子);(d) g-C3N4的高分辨率透射電子顯微鏡(HR-TEM)圖像;(e) g-C3N4@Ni泡沫的SEM圖像;(f)在不同電流密度下,g-C3N4@Ni泡沫、Ni泡沫和Cu電極上的Li成核超電勢118Fig. 8 (a) Crystal models for calculating the binding energy of a Li+ adsorbed on g-C3N4 and (b) the corresponding charge density difference (brown, purple, and green balls represent carbon atoms, nitrogen atoms, and Li atoms, respectively; yellow and light blue areas represent positive and negative charge differences, respectively); (c) crystal models for calculating the binding energy of a Li+ adsorbed on Ni (blue and green balls represent nickel atoms and Li atoms, respectively); (d) highresolution transmission electron microscope (HR-TEM) image of the g-C3N4; (e) SEM image of the g-C3N4@Ni foam;(f) Li nucleation overpotentials on g-C3N4@Ni foam, Ni foam, and Cu electrodes at different current densities,the nucleation overpotential is defined as the difference between the sharp tip voltage and the later stable mass transfer-controlled overpotential 118.
值得注意的是,Fan等119對鋰離子在不同過渡金屬摻雜銅集流體表面成核機制進行了理論研究。首先,選擇Zn、Ag、Au和Sn作為銅集流體表面的金屬摻雜原子,研究金屬摻雜銅表面與鋰金屬離子的相互作用關系;然后,通過第一性原理計算,從熱力學性質和動力學性質兩個方面探討了鋰離子在這些金屬摻雜銅表面上的沉積成核機理。計算模擬結果表明,摻雜不同金屬原子會引入吸附能梯度變化的沉積位點,提升鋰金屬離子的擴散性能和離子電導率。在不同的摻雜金屬原子中,鋅原子和銀原子摻雜的銅表面可以改善鋰離子的吸附和擴散性能。鋰離子沉積的梯度吸附能和動力學擴散特性都對鋰離子的初始成核過程產生影響。在設計鋰金屬無枝晶結構時應重點考慮這一點,這種摻雜的方法中,不需要使用超高表面積的三維多孔材料,且簡單易行。該從理論角度指導了實驗,進行了理論的篩選工作。關于Ag和Zn摻雜銅集流體表面的理論計算結果,均陸續得到相關實驗驗證120。
此外,還可以利用納米金剛石與鋰離子共沉積反應,調節充電過程中鋰的沉積行為,進而穩定鋰金屬電極121。與常規的銅材料相比,納米金剛石在鋰沉積過程中表現出親鋰的特性,降低了鋰均勻生長的擴散勢壘,從而為鋰?鋰對稱電池穩定循環創造條件。基于第一性原理的計算方法有助于揭示這一現象背后的機理。納米金剛石(110)晶面和Cu(111)晶面因具有最低表面能,被用于模擬鋰的吸附和擴散過程(圖9a)。由于過程中電子大量遷移,納米金剛石對鋰離子具有高達3.51 eV的結合能(比銅高出1 eV左右) (圖9b)。此外,在鋰金屬及SEI的各種組分(Li2O、Li2CO3、LiOH、LiF等)中,鋰離子在納米金剛石表面具有最低的擴散勢壘,這能夠誘導鋰的快速擴散、均勻沉積,從而避免枝晶的產生(圖9c,d)。
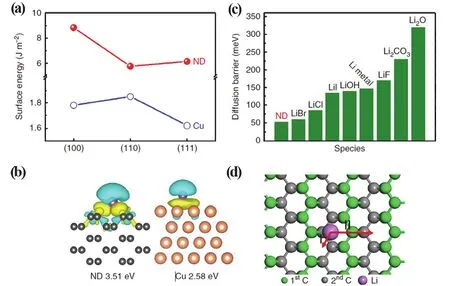
圖9 第一性原理計算描述納米金剛石表面上鋰離子電鍍過程121Fig. 9 First-principles calculations to describe Li ion plating behavior on nanodiamond surface 121.
利用界面能可以在熱力學上抑制枝晶生長,這是電極設計時值得關注的。Liu等122設計了一種Li + 11% (w) Sr合金負極,該合金電極在氟化電解質中可以形成富含SrF2的SEI。DFT計算模擬和實驗表征證明,富含SrF2的SEI與鋰金屬的界面能較大并且機械強度高,能夠促進鋰金屬橫向沉積,抑制枝晶生長,維持SEI的穩定性。該工作通過調節鋰金屬負極組成設計SEI,并利用界面能抑制枝晶生長,為研究提供了新的思路。Liu等120通過磁控濺射銅鋅合金的方法在銅箔集流體表面引入均勻的原子級鋅缺陷。由于鋰金屬可以首先與鋅形成固溶體,這使得鋰與集流體間的沉積界面能大幅降低,從而誘導鋰金屬在其表面均勻沉積。實驗證明該鋰金屬電極性能得到明顯改善。該工作為利用界面能抑制枝晶生長,提供了新的視角。Zhang等123利用氧化鋅和碳納米管構建出對鋰的親疏性具有梯度的保護層。其中,底層表現親理性,與鋰金屬緊密接觸,促進SEI均勻形成;頂層表現疏鋰性,并具有較高的機械性能和多孔結構,抑制枝晶生長,促進鋰離子擴散。該工作具有創新性。
6 固體電解質
近年來,應用固態電解質(Solid state electrolytes,SSEs)的全固態鋰電池以高能量密度、高穩定性安全性等優點引起人們關注。SSEs可以從根本上解決常規有機電解液易分解、金屬電極表面不平整等問題,減少許多安全隱患,并大幅提高電池的能量密度;但是依然存在界面傳輸阻礙大,枝晶生長難以控制,以及工作電流低等問題,其中以離子導電性問題最為突出84,124-128,有必要對此進行計算模擬,研究傳輸機制和傳輸速率。該部分將綜述計算模擬在SSEs中傳輸現象的應用,并重點關注SSEs與鋰電極間的相互作用,利用計算模擬的方法,對機理進行闡明,指導新型SSEs的設計和優化。
由于SSEs存在上述問題,深入認識SSEs中離子的傳輸機制顯得尤為重要。分子動力學(MD)方法可以很好地模擬鋰離子在SSEs中的擴散過程,是研究鋰離子擴散行為的有效工具。擴散系數是研究鋰離子擴散行為中的關鍵因素,根據Arrhenius定律,鋰離子的擴散系數

(其中D0為系數,Ea為鋰離子擴散能壘,kB為玻爾茲曼常數)。利用MD方法,可以計算鋰離子在不同組分不同狀態下的擴散系數,并根據Nernst-Einstein方程得到離子電導率。Fang等129用團簇離子代替基本離子,獲得新的鋰離子超導體Li3SBF4和Li3S(BF4)0.5Cl0.5,利用MD方法分別計算材料的鋰離子電導率。他們發現,Li3SBF4的室溫離子電導率估計值為0.1 mS·cm?1,擴散能壘低至0.210 eV,具有低形成能、高熔點,以及足夠的機械性能,與之結構類似的Li3S(BF4)0.5Cl0.5的混合相則具有約1 mS·cm?1的室溫離子電導率和0.176 eV的擴散能壘。該工作表明團簇離子可以有效提高離子電導率,為設計高離子電導率的SSEs提供了思路。Wang等130利用材料基因組方法對反鈣鈦礦Li3AX進行系統研究,其中使用MD方法評估相關化合物的鋰離子傳輸能力,計算其擴散系數。研究發現,Li6OSI2的空位傳輸和間隙傳輸的擴散能壘分別為0.26和0.22 eV,室溫下離子電導率分別為1.03和5.0 mS·cm?1,均優于Li3OCl (室溫下離子電導率分別為0.12和0.1 mS·cm?1)。鋰離子的傳輸路徑等相關機理研究也是MD方法在鋰離子傳輸行為研究中的重要應用之一。Wang等131利用MD方法對石榴石型固態電解質Li5Ta2O12中鋰的局部結構和動力學進行研究,從統計意義上分析鋰在四面體間隙、八面體間隙,以及三角形瓶頸中的分布位置和動力學行為,并指出其傳輸機理受溫度等環境條件影響很大。Smith等132利用MD方法對典型的玻璃態固態電解質75Li2S-25P2S5中鋰離子的傳輸進行研究,發現PS43?陰離子的定向運動與鋰離子的協同運動共同影響鋰離子的傳輸機制。其中陰離子以旋轉的方式與鋰離子協同運動,被稱為“明輪效應”,有利于室溫下離子的遷移。他們通過分析鋰離子傳輸與空間、時間、振動以及能量的關聯性,證實了明輪效應對鋰離子傳輸的貢獻。該工作揭示了玻璃態SSEs中鋰離子傳輸的具體細節。缺陷及晶界的存在會破壞SSEs晶體結構的周期性,進而影響鋰離子的擴散行為。之前,科研工作者對SSEs晶體結構中缺陷的研究主要關注在點缺陷和晶界上,Zhu等133發現了一種可以顯著影響離子傳輸的非周期特征,將其命名為“單原子層陷阱(SALT)”。SALT表現為單原子層二維缺陷導致的封閉環形,并在固態電解質Li0.33La0.56TiO3中大量存在。根據MD方法計算模擬發現,鋰離子不能在SALT中遷移,這意味著SALT包含的部分無法參與離子遷移過程,嚴重降低總電導率。該工作加深了人們對SSEs中非周期特征的認識。
計算模擬也被用于SSEs的優化設計。Xu等134通過構建陰離子亞晶格模型,利用DFT研究了陰離子電荷和晶格體積對面心立方(fcc)陰離子亞晶格中鋰離子占據形式和遷移活化能,并由此提出利用該模型設計、優化超離子導體的一般原則。該工作為高通量計算篩選鋰離子超導體提供了理論指導。Lee等135利用無過量鋰的銀-碳納米復合負極和硫化物固態電解質構建出高性能鋰金屬電池,并證明了復合負極在調節鋰金屬沉積、促進界面穩定方面的作用。這些工作都極大地豐富了人們對SSEs的認識,為進一步構建高性能SSEs奠定了基礎。
SSEs與鋰金屬電極的界面在保障全固態鋰電池的穩定和高效中十分重要,其中,鋰枝晶生長并且傳輸阻力大一直是需要解決的關鍵問題112-116。利用DFT等計算模擬的方法,可以深入揭示相關機理,指導全固態鋰電池的設計和優化;此外,高通量計算和機器學習等方法可以高效篩選材料,大大降低研發周期,有望成為材料科學新的研究方式。這里將主要圍繞電極界面,以無機固態電解質(ICEs)和有機聚合物固態電解質(SPEs)兩類主要的SSEs,從界面上的鋰枝晶生長和離子導電性兩方面,進行綜述。
ICEs雖然具有較高的機械強度,但由于其中存在晶界等缺陷,鋰枝晶仍會產生,嚴重情況下可導致兩電極短路、器件損壞。Mo等136將實驗與理論相結合,闡明了在具有代表性的LiBH4SSE電池中枝晶的形成原因,并對LiF抑制枝晶生長的機理進行研究。實驗表明,鋰離子在擴散過程中遇到電子,在SSE的晶界/孔隙中還原為零價態,不斷累積,最終導致短路(圖10e);而將不易傳輸電子的的LiF填入SSE的晶界/孔隙,可以有效抑制這一過程(圖10f)。理論計算進一步證實這一機理并揭示其相關細節。根據DFT計算鋰與LiBH4、LiF以及LiBH4?xFx之間的形成能(圖10a-d),可知其形成能均為正值且Li/LiF的形成能約為Li/LiBH4的三倍,這意味著鋰在其表面更難以形核,而Li/LiBH3.5F0.5的形成能有待提升。通過LiF填充方法,電池的臨界電流密度大大提高(原LiBH4的235%),可逆容量得到改善(TiS2?LiBH4-LiF?Li,137 mAh·g?1)。Han等137利用計時原位中子深剖技術檢測SSE中鋰離子濃度在充電過程中的動態變化,揭示鋰枝晶的形成機制。通過對LiPON、Li7La3Zr2O12(LLZO)和非晶Li3PS4這三種常見、具有代表性的SSEs進行了研究,發現在LiPON中難以觀測到鋰離子的濃度變化,而在LLZO和Li3PS4中觀測到鋰離子的大量沉積。研究表明,LLZO和Li3PS4的高電子電導率是鋰枝晶生長的主要原因,降低SSE的電子電導率而非提高其離子電導率是全固態鋰電池的設計關鍵。這些工作深入探究了界面上鋰枝晶的生長機理,并探究了抑制枝晶生長的方法,為構建穩定全固態鋰電池奠定基礎。
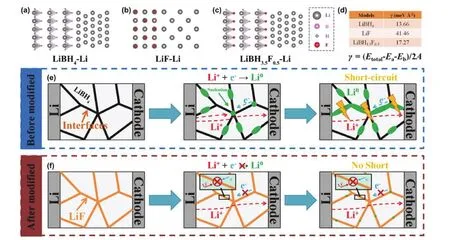
圖10 (a) LiBH4-Li、(b) LiF-Li和(c) LiBH3.5F0.5-Li的結構;(d)根據DFT計算得出的形成能;(e)未修飾的SSE中鋰枝晶生長機制;(f)使用LiF修飾后的SSE中鋰枝晶生長得到抑制136Fig. 10 The structure of (a) LiBH4-Li, (b) LiF-Li, and (c) LiBH3.5F0.5-Li; (d) formation energy of different models calculated by DFT. Schematic diagram of (e) Li dendrite formation in solid electrolyte before modified and(f) Li dendrite suppression mechanism in modified electrolyte system 136.
鋰金屬電極與SSE間巨大的界面電阻一直是限制全固態鋰電池發展的重要因素,過高的界面電阻會導致鋰金屬電池的過度極化。Duan等138為此制作出鋰-石墨復合電極,并將其熔融覆蓋于石榴型固態電解質Li6.5La3Zr1.5Ta0.5O12(LLZTO)的表面,兩者緊密接觸,界面阻礙降至11 Ω·cm2。實驗結果表明,石墨可能在鋰與LLZTO緊密接觸中起促進作用,而利用第一性原理計算發現,Li-C復合材料與LLZTO的反應能量為每原子?20至?100 meV,容易反應,而純Li和LLZTO顯示出較差的反應性質,從而證實了石墨對反應的促進作用。此外Huang等139將鋰和g-C3N4反應,由此制備的鋰電極也對石榴石SSE表現出很好的潤濕特性,從而降低界面阻礙,提高臨界電流密度。
除上述ICEs以外,一些有機聚合物116,如廣泛使用的聚環氧乙烷(PEO),也可以作為SSE,參與組成全固態鋰電池。這些有機聚合物固態電解質(SPEs)可以表現出與ICEs不同的性質,例如更低的界面阻礙、更好的柔韌性,同時也易被尖銳的鋰枝晶破壞140,141。因此,控制枝晶生長對SPEs而言更為重要,而且需要從不同于ICEs的角度考慮。Yan等142通過Mg3N2層修飾PEO,使鋰離子/電子分布均勻并提高機械強度,抑制枝晶產生。實驗發現,其中的Mg3N2會參與轉變為可以快速傳輸離子的Li3N和具有良好導電性的Mg,形成離子/電子混合中間層(MIECI),從而促進鋰離子/電子均勻分布。COMSOL仿真和XPS表征證明了MIECI的調節作用。此外,Li等143向PEO中引入Li10SnP2S12,在降低界面阻力,提高界面穩定性方面取得很好的結果;Wan等144向多孔聚酰亞胺(PI)膜中填充PEO/LiTFSI復合物制備聚合物-聚合物固態電解質,使電池具備優異的循環性能,并且可以承受彎曲、切割和針刺等極端狀態;Zhao等145利用含鋁陽離子在電池內部引發醚分子開環聚合來制備SPE,發現與鋰電池的各組件接觸良好,表現出高離子電導率(> 1 mS·cm?1)、低界面阻抗、均勻沉積和高庫侖效率(> 98%,300次循環)。
許多科研工作者將機器學習應用于加快研發高性能SSEs。Sendek等146首先從大量含鋰固體中篩選出穩定、電子電導率低及廉價的一萬多份樣本,應用機器學習對其關于離子電導率分類,以幫助確定可快速導通鋰離子的結構,從而將樣本歸結為21種具有潛在應用價值的結構,其中多數未曾經過實驗檢驗。該工作還發現,對離子電導率的預測需要綜合多因素建立復雜的預測模型。Zhang等147在材料數據較少的情況下,利用機器學習成功區分了室溫下傳導鋰離子的良性材料和不良材料,進而篩選出16種室溫離子電導率高于10?4S·cm?1的固態鋰離子導體,部分甚至超過了10?2S·cm?1。新發現的固態鋰離子導體具有完全不同于已知導體的結構和組成。Kahle等148、He等149將DFT和MD與高通量計算相結合,對鋰離子良性導體進行計算模擬和篩選,取得很好結果。這些工作對應用機器學習、高通量計算篩選指定性能的材料有重要的指導意義。Harada等150利用人工智能研究SSE最優摻雜劑的種類和濃度。研究者們通過向固態電解質材料NASICON型磷酸鋯鋰摻雜鈣離子和釔離子,制備了47種樣品材料,評估發現鋰離子電導率對成分的依賴十分復雜,僅依靠經驗和直覺來尋找最佳成分十分困難。為此,他們利用貝葉斯優化方法進行實驗尋找最優結果,確認僅需1/3的樣品數據即可以高于99.9%的概率找到最優解。該工作可以啟發應用機器學習對SSEs成分進行優化設計。
7 潛在研究方向
目前,理論模擬在鋰電極領域取得了極大進展,為認識電解液與金屬電極的界面反應、改進SEI、構建復合電極材料提供了許多新的見解。但由于各方面困難,許多基礎的、重要的問題仍未解決:一方面,一些基礎問題,如鋰的不均勻沉積、界面反應,在理論層面仍沒有探索清楚,實驗觀測結果也有待在原子水平上揭示背后的機理;另一方面,人們雖然已經開發出許多有效方法以抑制鋰枝晶生長,特別是引入新型框架材料和界面材料,但只有更深入地認識這類材料,才能夠合理地設計負極。
該領域的潛在研究方向如下:
(1)開發更適用的計算模擬方法。對于復雜的鋰化界面,過渡態理論和分子動力學方法都面臨著許多問題,故急需尋找更加適用于鋰電極的方法。可以將其他鋰電池系統的模擬方法應用于金屬負極的研究。例如Chan等151基于第一性原理,開發出一種關于鋰插入移除的算法,用于模擬晶體硅中鋰化和脫硫過程。這一工作在動力學層面為后續深入研究鋰離子的生長、成核機制提供了啟發性思路。鋰枝晶在無缺陷界面上的生長特性,也可以新算法的設計有所啟發,解決鋰動態生長的相關問題。
(2)結合先進表征技術。應用先進實驗表征技術可以驗證計算模擬結果、啟發新的計算模擬方法,是未來深入研究的重要方式。He等152通過將原子力顯微鏡的懸臂耦合到環境透射電子顯微鏡中的固體開孔裝置中,可以直接觀測鋰枝晶的形核生長;Yuki等153利用透射電子顯微鏡和機器學習,開發出了可在納米水平實時觀察塊狀和薄膜狀全固態電池內部的鋰離子運動的技術,以此研究充放電過程中鋰離子的擴散。這些先進觀測技術的結果有助于認識機理,為計算模擬提供思路。
(3)深入研究鋰成核沉積機理。在鋰離子沉積過程中,枝晶的形成是一個自發的熱力學過程。為控制鋰金屬電極的表面形態,抑制鋰枝晶生長,應深入研究影響鋰成核動力學過程和沉積形態的關鍵因素。目前,Jana等154基于擴散熱力學和電沉積動力學,對不同條件下鋰離子沉積情況進行計算模擬,提出3種生長機制和6種枝晶形態,與實驗相符,形成一套完整的體系。已提出的“鋰鍵”,也影響著電解質中的含鋰團簇和溶劑化結構以及鋰在電極表面的沉積155,或許可以為相關機理研究提供思路。
(4)合理設計鋰電極。實驗反復證明,合理設計界面是保護金屬陽極最有效的方法之一,目前已相繼發現不同類型的界面材料。例如,Tu等156通過在鋰金屬電極上沉積電化學活潑的金屬(如Sn、In或Si)來制備混合型電極,使電極材料具有良好的循環性能;Gao等157在分子級別上設計SEI,材料表現出優異的鈍化性質、均質性和機械強度。超分子橡膠也可應用于設計具有自修復能力的人工SEI158。這類高粘彈性、可自愈材料可以防止SEI產生縫隙或針孔,促進鋰離子的均勻沉積,但帶來離子電導率下降。這或許是設計人工SEI的新思路。除此之外,為鋰金屬負極構建穩定的基體框架也是一個方向。對此,理想的基體表面對鋰離子應具有較高的親和能力,而基體也應具有足夠的強度和傳導性能。到目前為止,人們已經設計了各種較為合適的基體框架。例如類珊瑚銀包覆的碳纖維基體159、三維泡沫鎳基體160、LiAlF161和SiOx162等等。
(5)綜合優化電池系統。復雜的界面反應以及成核沉積等現象說明,鋰電池系統中的多種因素,以復雜的關系和效應,共同影響著鋰負極的工作性能。因此,有必要采用新方法來實現整體優化。例如,鋰化過程會極大地影響基體材料的結構和性能,這就要求對基體材料的結構進行識別。進一步,利用結構預測的方法,將鋰離子遷移能力和抑制枝晶能力作為關鍵指標,系統地研究相關問題,從而在未來實現對基體材料的理論優化。目前,有研究結果表明鋰金屬負極的厚度等設計參數對電池的循環壽命和能量密度有復雜的關系163,164,Severson等43也利用機器學習方法對鋰電池的循環壽命進行預測,揭示系統中復雜的非線性關系。這意味著,為設計合理高效的鋰電池,需要將電池的各要素綜合考慮。機器學習可以作為解決上述問題的有力工具。
猜你喜歡
艦船科學技術(2022年16期)2022-09-22 02:15:00
北京航空航天大學學報(2021年6期)2021-07-20 07:23:54
當代陜西(2020年13期)2020-08-24 08:22:02
制造技術與機床(2017年5期)2018-01-19 02:49:17
制造技術與機床(2017年11期)2017-12-18 06:47:29
金秋(2017年4期)2017-06-07 08:22:16
蘇州科技大學學報(自然科學版)(2017年1期)2017-03-20 15:25:18
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
