SOTOS綜合征NSD1基因新發錯義突變1例并文獻復習
2021-03-18 06:08:24
精準醫學雜志 2021年1期
(青島大學附屬醫院新生兒科,山東 青島 266003)
SOTOS綜合征為一種先天的過度生長性疾病,以常染色體顯性遺傳,主要表現為特殊面容、學習障礙以及生長過度,發病率約為1/14 000[1]。超過90%的患者存在NSD1基因異常,臨床上癥狀與SOTOS綜合征高度相似的非NSD1基因突變患者,已被證明攜帶APC2和NFIX基因突變[2]。本文報道1例NSD1基因新發錯義突變患兒,并結合相關文獻進行復習,探討基因型與表型的關系,為提高臨床醫師診斷該病提供經驗。
1 臨床資料
對1例5月齡SOTOS綜合征男嬰的臨床資料進行回顧性分析,并對患兒及父母進行全外顯子組基因檢測。獲父母知情同意和醫院倫理委員會批準后,采集患兒及父母外周靜脈血各2 mL,EDTA抗凝,以QIAampDNA Blood Mini試劑盒(德國Qiagen GmbH公司)按廠家說明提取基因組DNA。將提取的基因組DNA片段化和擴增純化以后與適配器相連接,由IDT XGen外顯子研究小組(IDT,Lowa,美國)的探針捕獲約19 396個靶向基因,構建DNA文庫。DNA文庫再經擴增和純化以后,在NovaSeq 6000測序儀(Illumina,San Diego,美國)上采用Burrows-Wheeler Aligner(v.0.5.9-r16)軟件進行測序,測序結果與UCSChg19人類參考基因組序列進行比對,使用PriVar Toolkit工具進行數據注釋后,確定有臨床意義的變異體,再用PCR方法驗證檢測到的變異,并在3500XL遺傳分析儀上對獲得產物進行直接測序。通過SIFT、PROVEAN PREDICTION、Mutation Taster、Polyphen2等生物信息學軟件預測致病性。
2 結 果
2.1 臨床表現及體征
患兒,男,5月齡,因“發育遲滯”就診。系第2胎第2產,胎齡40+4周,順產出生,出生時體質量為3 990 g,Apgar評分10分,無窒息史。母親孕期體健。自出生以來,喂養困難,吮吸差,哭聲直,現仍不能逗笑,不會咿呀作語,不能追視。出生后10 d因“新生兒高膽紅素血癥、新生兒肺炎”就診當地醫院,最高時總膽紅素為389.9 μmol/L,住院10 d好轉出院。家族史:父母體健,否認為近親結婚,否認有家族遺傳病史,患兒有1個3歲姐姐,體健。查體:頭圍49.2 cm,身高71 cm,體質量7 kg。前囟未閉,約2 cm×2 cm大小,平軟。巨顱,顱骨重疊,前額突出,下頜尖長,高腭弓,下牙齦凹凸不平(圖1A、B),萌牙2顆。患兒雙耳廓外形基本正常,雙耳(長約6 cm)、雙手(長約8 cm)、雙腳(長約11 cm)較同齡兒明顯偏大,雙側指掌紋無異常,心、肺、腹查體未見異常。雙手拇指內收,四肢肌張力高,病理征及腦膜刺激征陰性。抬頭差,豎抱頭不穩,視聽追視無反應,不會逗笑,不能與人互動,哭聲直,發音單一,不會咿呀作語,無主動抓握意識,不可獨坐。精細運動、大運動及語言發育均落后同齡兒。
2.2 實驗室檢查結果
血常規、生化、心臟以及泌尿系統超聲檢查均未見有明顯異常;顱腦MRI檢查顯示前后徑增大,腦溝深,腦回增寬,雙側基底節區T1WI稍高信號,雙側內囊后肢T1高信號,雙側腦室體部異常信號(圖1C~E)。
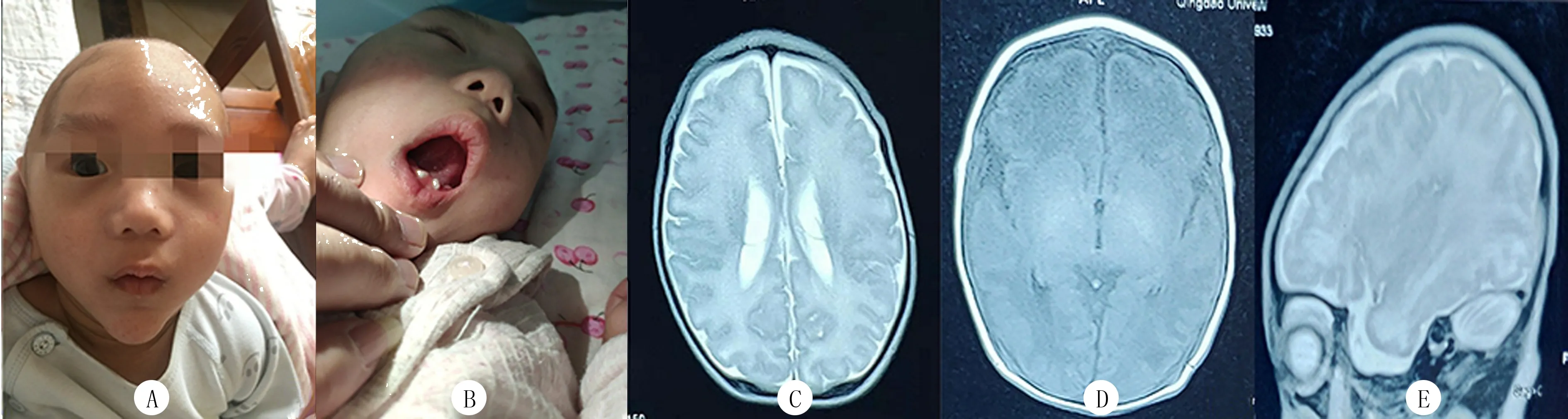
A:患兒的前額突起,下頜尖長;B:患兒下牙齦凹凸不平,牙列不齊;C~E分別為T1軸位、T2軸位、T1矢狀位顱腦MRI圖像
2.3 基因檢測結果
基因檢測結果顯示,該患兒5號染色體5q35區出現NSD1基因區域錯義突變c.5791T>C,使該位點的半胱甘酸轉變為精氨酸(p.Cys1931Arg)。在HGMD數據庫中并未見相關文獻報道;dbSNP147數據庫、ESP6500siv2_ALL數據庫和千人基因(1000g2015aug_ALL)數據庫均未見收錄。其父母樣本中未檢測到該變異,因此推測其為新發突變。患兒、患兒父親、患兒母親的基因檢測結果見圖2。
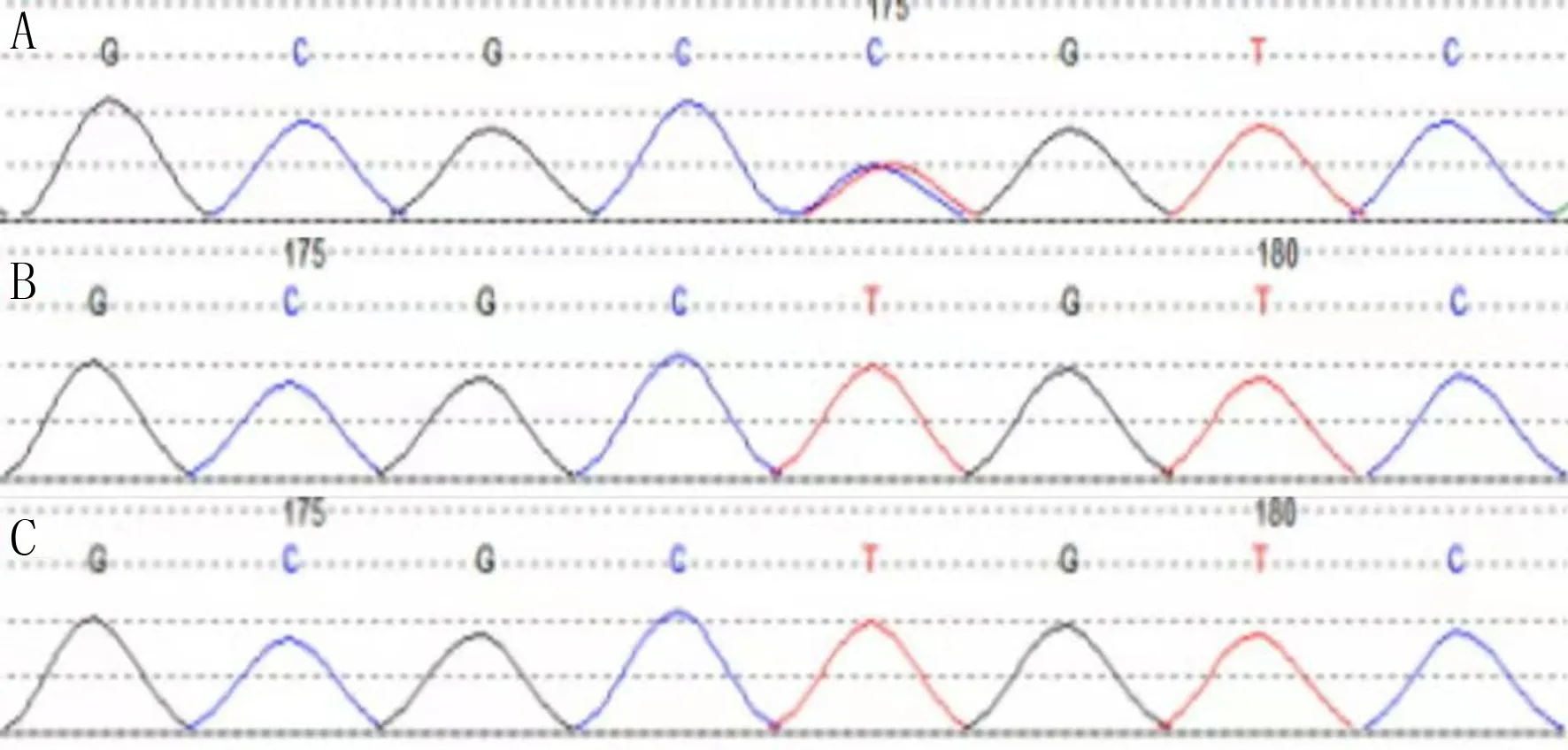
A、B、C分別為患兒、患兒父親、患兒母親
3 討 論
3.1 SOTOS綜合征患者的臨床表現
超過90%的SOTOS綜合征患者具有以下臨床特征[3]:①特殊面容,如前額凸出、鼻梁低平、輕度斜視、高腭弓、眼距增寬、眼裂下斜、小下頜、尖下巴及額頂區毛發稀疏等;②學習障礙,如早期發育遲緩,輕度至重度智力障礙;③過度生長,如在青春期前,尤其1~6歲,頭圍或身高大于正常同齡兒第97百分位,或平均值的2個標準差;在青春期后,身高增長趨于正常,但大頭畸形非常典型;④骨齡超前。其他臨床表現還有腦室擴大、早期喂養困難、新生兒高膽紅素血癥、肌張力減退、新生兒低血糖、皮膚錯構瘤、脊柱側彎、關節過度松弛、扁平足、癲癇、便秘、腫瘤、淋巴水腫、先天性心臟病(動脈導管未閉、室間隔缺損及房間隔缺損等)及泌尿生殖系統異常(膀胱輸尿管反流、隱睪、多囊腎)等[4-9]。成年SOTOS綜合征患者有一個獨特的面部特征,呈方形且突出的下頜。一般成年患者都很健康,僅少數患者會出現淋巴水腫、牙列不全、聽力喪失、肢體攣縮和震顫等臨床癥狀[10]。此外,SOTOS綜合征患兒易合并孤獨癥、注意力缺陷與多動障礙、焦慮行為以及語言障礙等[11-15]。
本研究報道的該例5月齡的患兒,存在典型的SOTOS綜合征臨床表現,且顱腦MRI信號異常,可能存在神經系統異常。因家屬拒絕骨齡檢查,尚不能明確是否存在骨齡超前。該患兒經基因檢測,發現5號染色體q35.3區出現NSD1基因區域錯義突變c.5791 T>C,使該位點的半胱甘酸轉變為精氨酸(p.Cys1931Arg),生物信息學軟件預測其致病性很高,此突變點既往未有文獻報道。結合基因檢測結果,患兒符合SOTOS綜合征診斷。
3.2 SOTOS綜合征患者基因型與表型的關系
SOTOS綜合征主要是由于NSD1基因單倍劑量不足所致,包括NSD1基因內突變(錯義突變、無義突變、移碼突變等)和5q35微缺失[1,16]。SOTOS綜合征基因型在種族之間存在差異,于歐美國家中,NSD1基因突變型占80%~85%,5q35微缺失型占10%~15%;而在日本患者中,5q35微缺失型占50%以上[1]。本研究中的患兒NSD1基因存在未見報道的雜合突變(c.5791 T>C),臨床表現與文獻報道相似。中國內地共報道了17例SOTOS綜合征患兒,有15例經基因檢測確診,其中NSD1基因突變型8例(4例錯義突變,3例移碼突變,1例無義突變),占47%(8/17);5q35微缺失型7例,占41%(7/17),均為散發病例,未見有家族性報道[17-25]。中國香港地區報道了36例SOTOS綜合征,26例患兒NSD1基因異常,其中NSD1基因內突變型23例,占64%(23/36),5q35微缺失型只有3例,僅僅占8%(3/36)[26]。按照我國人口數量,報道的樣本量過小,不能明確我國患者的基因型,說明我國對此類罕見病的臨床認識不足。有研究報道5q35微缺失型與NSD1基因突變型之間存在表型差異,與基因突變型相比,5q35微缺失型患者更容易出現學習困難及心臟、腎臟異常等問題,而過度生長現象不明顯[27]。在我國報道的17例患兒中,大部分存在特殊面容、過度生長及智力障礙等臨床表現,幾乎所有的患兒顱腦MRI顯示腦室擴大、腦發育不良,部分患兒合并骨齡超前、低糖血癥、腭裂、新生兒喂養困難、新生兒黃疸等。本研究報道的基因內突變型患兒臨床表現與國內報道的相符,早期合并喂養困難及新生兒黃疸,無心臟及腎臟異常。在國內報道的17例患兒中,有4例患兒合并先天性心臟病或腎臟異常,皆為5q35微缺失型,與既往文獻報道相符,可能微缺失型患兒更易合并心臟及腎臟異常。康路路等[17]報道了3例NSD1基因突變型患兒中,有1例患兒無特殊面容及過度生長。趙敏[20]報道了3例SOTOS綜合征患兒,2例NSD1基因突變型無過度生長。孫碧君等[25]報道了2例微缺失型SOTOS綜合征新生兒,1例出生時即表現為過度生長,另1例在隨訪至45個月時出現過度生長,與國外研究有差異,可能與我國樣本量較小有關系。BOU-ASSI等[28]研究報道了1例以新生兒皮膚松弛癥作為首發癥狀的SOTOS患兒。SIO等[29]報道了首例SOTOS綜合征合并先天性巨結腸患兒。SOTOS綜合征新生兒期的癥狀更加不典型,合并罕見臨床癥狀時,可考慮存在基因異常。相關研究表明,高胰島素血癥是新生兒期SOTOS綜合征患兒的特征性癥狀[30-31]。GRAND等[32]報道了7例合并高胰島素血癥的SOTOS綜合征患兒。在高胰島素血癥或低血糖的背景下,即使無過度生長表現,也可以考慮此病[33],并應進行早期的基因診斷,以改善患兒預后。LACCETTA等[34]報道了1個家系的SOTOS綜合征,該家族3代成員攜帶相同突變位點,先證者具有典型SOTOS綜合征表現,母親與外祖父僅身高大于正常同齡人第97百分位,無其他異常。韓國報道了1對同卵雙生子患兒,2名患兒均為NSD1基因無義突變,先證者存在癲癇發作、脊柱側凸、腦室擴大、注意力缺陷、多動障礙等異常的臨床表現,而其胞弟并無上述表現,僅有SOTOS綜合征的典型臨床表現[35]。因此,突變位置與臨床表型之間沒有絕對的相關性,同一突變位點也可以表現出不同的臨床特征。SOTOS綜合征相關的臨床特征,可能與基因型無關,具體機制尚不清楚,有待進一步研究。
3.3 SOTOS綜合征的產前診斷及隨訪
如果產前超聲顯示胎兒生長過度、頭顱畸形、羊水過多且合并其他異常,如泌尿系統異常、中樞神經系統異常、頸部透明帶增厚和異常的母體血清篩查結果,應考慮此病,并應早期進行基因檢測,有助于遺傳診斷和咨詢[36-37]。目前此病尚無特殊治療方法,兒科定期隨訪很重要,以便早期處理并發癥,提高患兒預后。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中國生殖健康(2020年2期)2021-01-18 02:51:26
家庭醫學(下半月)(2019年9期)2019-10-12 08:04:06
家庭醫學(下半月)(2019年8期)2019-09-25 09:02:00
小學生導刊(2018年13期)2018-06-29 03:49:00
媽媽寶寶(2017年3期)2017-02-21 01:22:12
海峽科技與產業(2016年3期)2016-05-17 04:32:12
