X連鎖低磷酸鹽血癥性佝僂病家系1例報(bào)告并文獻(xiàn)復(fù)習(xí)
2021-03-18 06:08:24
精準(zhǔn)醫(yī)學(xué)雜志 2021年1期
(青島大學(xué)附屬醫(yī)院,山東 青島 266003 1 內(nèi)分泌代謝科; 2 內(nèi)科學(xué)教研室; 3 醫(yī)務(wù)科)
X連鎖低磷酸鹽血癥性佝僂病(XLHR)一般始發(fā)于嬰兒期,為X連鎖顯性遺傳,是最常見的遺傳性佝僂病。該病發(fā)病機(jī)制是由于磷酸鹽調(diào)節(jié)基因(PHEX)突變影響了腎小管對(duì)磷的重吸收,從而導(dǎo)致尿磷排泄增多,血清磷酸鹽水平下降。其臨床特點(diǎn)主要表現(xiàn)為身材矮小,發(fā)育遲緩,下肢嚴(yán)重彎曲,如弓形腿,部分患者常伴有牙齒畸形。本病在臨床上易誤診,傳統(tǒng)治療以口服磷酸鹽補(bǔ)充劑聯(lián)合活性維生素D類似物(阿法鈣化醇或骨化三醇)為主,但是長(zhǎng)期接受該療法的成人患者容易引發(fā)甲狀旁腺功能亢進(jìn)和腎鈣化。因此本文針對(duì)我院收治的一例XLHR家系的臨床資料情況進(jìn)行分析,并結(jié)合文獻(xiàn)復(fù)習(xí),探討產(chǎn)前及產(chǎn)后的早期診斷對(duì)XLHR幼兒患者治療及預(yù)后的意義。
1 臨床資料
患者,女,36歲,自出生即檢查出血清磷酸鹽水平偏低。該患者幼時(shí)即有膝內(nèi)翻表現(xiàn),無其他伴隨癥狀,曾接受過磷酸鹽治療,劑量未知。7歲時(shí)接受膝內(nèi)翻矯形手術(shù),術(shù)后不再服用磷酸鹽,無其他不適。于2019年3月21日因左髖疼痛、久坐以及活動(dòng)后疼痛加劇來我院就診,患者左髖屈伸檢查正常,后轉(zhuǎn)診至內(nèi)分泌科。入院檢查:血清堿性磷酸酶(ALP)117.00 U/L,血磷0.63 mmol/L,25羥維生素D3(25-OH-D3)20.00 μg/L,血鈣2.16 mmol/L,甲狀旁腺激素(PTH)1.55 μg/L,β-膠原降解產(chǎn)物0.22 μg/L,血肌酐35.00 μmol/L。腰椎正側(cè)位、骨盆正側(cè)位、左股骨正側(cè)位X線檢查顯示骨盆骨密度降低,雙側(cè)骶髂關(guān)節(jié)、髖關(guān)節(jié)未見明顯異常;左中股骨不規(guī)則,腰椎向右微彎曲。骨密度測(cè)定示腰椎T值0.62,左股骨T值-2.53。
患者兒子,2歲,12.6 kg。2019年2月1日于我院產(chǎn)科出生,其骨代謝相關(guān)指標(biāo)檢查結(jié)果顯示,血磷為1.46 mmol/L,血鈣為2.05 mmol/L,25-OH-D3為18.20 μg/L,PTH 0.51 μg/L,其他檢查如血常規(guī)等未見明顯異常。
患者母親,58歲。2019年5月29號(hào)于我院門診進(jìn)行骨代謝相關(guān)指標(biāo)檢查示:血磷 0.67 mmol/L,血鈣2.39 mmol/L,血肌酐60.00 μmol/L,血鎂0.90 mmol/L,其他指標(biāo)未見明顯異常。
由于患者未進(jìn)行產(chǎn)前遺傳病檢查,為明確診斷、指導(dǎo)治療,于患者兒子出生2 d后同時(shí)采集患者及其兒子、母親的血液標(biāo)本進(jìn)行骨骼發(fā)育異常相關(guān)基因檢測(cè),結(jié)果顯示三者的磷酸鹽調(diào)節(jié)內(nèi)肽酶基因(PHEX)上均有一相同移碼突變 (c.1097delT)(圖1),該突變位點(diǎn)以前未見相關(guān)文獻(xiàn)報(bào)道,因此初步診斷該患者家系為XLHR家系。
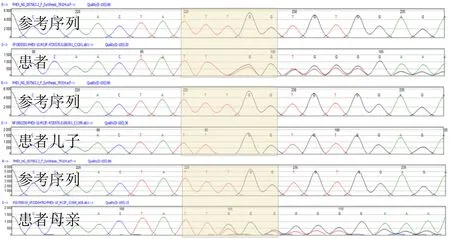
圖1 患者及其兒子、母親的PHEX基因突變位點(diǎn)圖
基因檢測(cè)后即給予患者兒子口服磷酸鹽進(jìn)行治療,每日5次,每次10 mL;鈣三醇丸每日1次,每次0.25 μg,患者未予藥物治療。1月后患者因左髖疼痛就診后給予磷酸鹽治療,每日5次,每次30 mL;鈣三醇丸每日1次,每次0.5 μg。患者母親未予藥物治療。
3月后復(fù)查,患者血磷 0.76 mmol/L,25-OH-D323.50 μg/L,其他指標(biāo)未有明顯改善,自述髖部疼痛未有明顯改善。患者兒子血鈣2.18 mmol/L,其他指標(biāo)未有明顯改善,下肢X線檢查顯示下肢略彎曲,呈“O”形。患者母親未接受復(fù)查。根據(jù)復(fù)查結(jié)果,將患者的治療計(jì)劃調(diào)整為磷酸鹽每日5次,每次30 mL;鈣三醇丸每日3次,每次0.25 μg。患者自述兒子無不良反應(yīng),故保持原治療計(jì)劃,并保持定期隨訪。
2 討 論
XLHR是一種由腎臟磷酸鹽消耗引起的低磷酸鹽血癥性疾病[1]。在臨床上,XLHR的主要診斷依據(jù)是患者的發(fā)病年齡、臨床癥狀、血清磷酸鹽濃度是否降低、ALP濃度是否升高、PTH及尿鈣是否正常,以及患者家系中是否存在有PHEX基因突變等[2]。本例患者幼年時(shí)即出現(xiàn)膝內(nèi)翻,成年后以髖關(guān)節(jié)骨痛為主要癥狀,骨骼發(fā)育異常相關(guān)基因檢測(cè)發(fā)現(xiàn)患者及其母親、兒子均存在相同PHEX基因突變,故診斷為XLHR家系。
X連鎖顯性遺傳是低磷酸鹽血癥性佝僂病最常見的可遺傳形式,遺傳發(fā)生率大約為1/20 000,約占HR的80%[2]。研究發(fā)現(xiàn),XLHR與人類X染色體短臂上的PHEX基因的突變密切相關(guān)。PHEX基因位于人類染色體Xp22.1上,包含22個(gè)外顯子,編碼749個(gè)氨基酸。該基因主要表達(dá)于成骨細(xì)胞中[3],編碼可降解局部整聯(lián)蛋白結(jié)合配體、N-連接糖蛋白和骨橋蛋白的酶[4],還可以抑制血漿中磷脂酰肌醇和成纖維細(xì)胞生長(zhǎng)因子(FGF23)的水平[5],PHEX基因突變可能導(dǎo)致內(nèi)肽酶產(chǎn)生不足和腎臟磷酸鹽排泄增加[6]。
目前關(guān)于XLHR的發(fā)病機(jī)制普遍認(rèn)為是由于PHEX基因功能性突變,通過FGFR途徑增加了FGF23的表達(dá),從而影響腎小管對(duì)磷的重吸收[7-8]。另外,細(xì)胞外基質(zhì)磷酸糖蛋白相關(guān)蛋白也可以影響FGF23的表達(dá)并調(diào)節(jié)腎臟磷酸鹽的代謝[9]。還有研究表明,PHEX蛋白可以通過上調(diào)蛋白轉(zhuǎn)化酶枯草桿菌蛋白酶/kexin-type2來裂解FGF23,并促進(jìn)PHEX-人牙本質(zhì)基質(zhì)蛋白1-整合素復(fù)合物的形成,從而降低FGF23的水平[10],但是由于缺乏蛋白質(zhì)之間直接相互作用的證據(jù),這種說法目前仍然存在爭(zhēng)議[11]。本例患者及其母親、兒子的PHEX基因第10外顯子中均發(fā)現(xiàn)有一處未見相關(guān)報(bào)道的突變(c.1097delT),該移碼突變會(huì)導(dǎo)致其編碼的蛋白質(zhì)從第366位異亮氨酸開始發(fā)生移碼并過早終止翻譯,導(dǎo)致編碼出的蛋白質(zhì)截短體失去正常蛋白功能。盡管人類基因突變數(shù)據(jù)庫(kù)已報(bào)告了400多個(gè)PHEX基因突變[12],但PHEX基因突變后所編碼的蛋白尚不清楚,因此c.1097delT突變對(duì)XLHR的因果表型效應(yīng)尚需進(jìn)一步研究[13]。
XLHR在幼兒患者中很難做出診斷,極容易被誤診為維生素D缺乏癥,只有當(dāng)患兒出現(xiàn)生長(zhǎng)發(fā)育遲緩、步態(tài)異常、顱骨畸形和骨痛等臨床表現(xiàn)時(shí),通過生化檢查排除了維生素D缺乏癥,再進(jìn)行基因檢測(cè)后才能確診[14-15]。該患者兒子剛出生時(shí)骨代謝指標(biāo)正常,依據(jù)骨代謝相關(guān)基因檢測(cè)發(fā)現(xiàn)了PHEX基因突變,并參考其母親及外祖母的基因檢測(cè)結(jié)果,被確診為XLHR。從生化角度看,XLHR患兒的特征是ALP升高,1,25-(OH)2-VitD3水平過低,血鈣和25-OH-D3水平正常[16]。但是,新生兒正常的血清磷酸鹽水平是成人的2倍,僅依靠骨代謝指標(biāo)很難明確診斷,并且新生兒可有一定程度的足內(nèi)翻,因此,患兒在2歲以內(nèi)經(jīng)常因忽視了骨骼畸形而錯(cuò)失治療良機(jī)。此外,部分患兒常常以牙周膿腫為首發(fā)癥狀[17],因此部分父母可能會(huì)依據(jù)患兒的牙周癥狀誤認(rèn)為牙齒疾病,從而延誤了XLHR的診治。
由于大多數(shù)PHEX突變發(fā)生在87%的家庭病例和72%的散發(fā)病例中[18-19],因此僅依靠家族史很難推斷出新生兒是否患有XLHR,只有通過基因檢測(cè)并參考臨床表型才能確診。LIAO等[20]對(duì)2例患有XLHR的孕婦進(jìn)行了基因檢測(cè),在1例患者家系中發(fā)現(xiàn)了一個(gè)新的錯(cuò)義突變c.1721T>A,其母親和舅舅也有相同突變,通過從其羊水中提取的胎兒DNA測(cè)序顯示,胎兒的基因位點(diǎn)在家族突變位點(diǎn)處為純合子峰,初步判斷胎兒出生后可能不會(huì)患有該病,嬰兒出生后1個(gè)月進(jìn)行基因檢測(cè)證實(shí)其未患有XLHR;另一例孕婦患者產(chǎn)前未行胎兒基因檢測(cè),分娩后第2 天對(duì)新生兒進(jìn)行基因檢測(cè),發(fā)現(xiàn)其攜帶與母體相同的突變,從而得到了及時(shí)治療。
目前,XLHR的傳統(tǒng)治療方法主要是口服活性維生素D類似物(阿法鈣化醇或骨化三醇)聯(lián)合磷酸鹽補(bǔ)充劑[17],但是用藥劑量應(yīng)根據(jù)血清中PTH、ALP和尿液中鈣/肌酐的濃度隨時(shí)調(diào)整,以降低嚴(yán)重副作用發(fā)生的風(fēng)險(xiǎn)[21]。臨床研究表明,聯(lián)合用藥可以改善30%~60%的患者的骨痛癥狀[22],可以在1年內(nèi)有效降低ALP濃度[17],減少由牙本質(zhì)礦化等引起的牙周膿腫[23]。但需注意該療法有引起甲狀旁腺功能亢進(jìn)和腎鈣化的風(fēng)險(xiǎn)[24]。對(duì)于早期確診的患兒,早期聯(lián)合用藥雖然有一定的效果,但仍然有24%~65%的患兒會(huì)出現(xiàn)下肢畸形,需要進(jìn)行矯形手術(shù)[25]。
XLHR的新式療法中研究最多的是應(yīng)用人源化的FGF23單克隆抗體(Burosumab)進(jìn)行治療,其可通過抑制FGF23來改善磷酸鹽轉(zhuǎn)運(yùn)蛋白的表達(dá),從而提高患者血清磷酸鹽水平[26]。在兩項(xiàng)分別針對(duì)幼兒和成人的臨床試驗(yàn)中已經(jīng)證實(shí),Burosumab可有效提高患者血清磷酸鹽水平,且患者僅有輕至中度不良反應(yīng),如頭痛及背痛等[27]。目前歐洲已經(jīng)批準(zhǔn)Burosumab為一種新的替代療法,可用于歐洲1歲以上的患兒[14],但其是否會(huì)引起甲狀旁腺功能亢進(jìn)和腎鈣化,尚不清楚,因此目前仍未被應(yīng)用于常規(guī)治療。
XLHR在疾病發(fā)展過程中易引起腎臟鈣化、聽力障礙、風(fēng)濕病和心血管系統(tǒng)疾病等并發(fā)癥,大大降低患者的生活質(zhì)量。研究證實(shí),接受早期治療的患兒,身高標(biāo)準(zhǔn)差評(píng)分要優(yōu)于出現(xiàn)骨骼癥狀后再接受治療的患兒[28]。在出生后18個(gè)月內(nèi)接受磷酸鹽和骨化三醇聯(lián)合治療的患兒,相較于18個(gè)月內(nèi)未經(jīng)治療的患兒生長(zhǎng)速度有所提高[29]。因此,提倡對(duì)發(fā)現(xiàn)有XLHR家族史的患者進(jìn)行婚前診斷,對(duì)有生育需求的患者進(jìn)行產(chǎn)前的胎兒基因檢測(cè)及產(chǎn)后的新生兒基因檢測(cè),這對(duì)患兒的早期診斷、治療及預(yù)后都具有重要意義。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫(yī)科大學(xué)學(xué)報(bào)(2021年2期)2021-03-29 05:31:08
現(xiàn)代臨床醫(yī)學(xué)(2021年1期)2021-01-26 00:56:02
中國(guó)生殖健康(2020年2期)2021-01-18 02:51:26
小學(xué)生導(dǎo)刊(2018年13期)2018-06-29 03:49:00
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
