蜂房的質(zhì)量標(biāo)準(zhǔn)研究
2021-09-19 08:03:08解紅霞郝偉亮趙炳聰賈福群
內(nèi)蒙古醫(yī)科大學(xué)學(xué)報 2021年4期
關(guān)鍵詞:標(biāo)準(zhǔn)
解紅霞,郝偉亮,趙炳聰,賈福群
(1.內(nèi)蒙古醫(yī)科大學(xué)附屬醫(yī)院藥劑部,內(nèi)蒙古 呼和浩特 010050;2.內(nèi)蒙古醫(yī)科大學(xué)藥學(xué)院)
蜂房是胡蜂科昆蟲果馬蜂[polistes olivaceous(DeGeer)]、日本長腳胡蜂(polistes japonicas saussure)或異腹胡蜂(parapolybia varia fabricius)的巢[1]。目前對蜂房的研究多集中于化學(xué)成分及藥理作用方面。在中醫(yī)臨床上,蜂房常用于治療肺癌[3,4]、類風(fēng)濕關(guān)節(jié)炎[5,6]等疾病。姚娓[7]等通過實(shí)驗(yàn),驗(yàn)證了蜂房提取物具有抑制小鼠移植性肝癌H22腫瘤的作用。王輝[8]等以蜂房組合黃芪、蘇木及其他藥材進(jìn)行配伍,發(fā)現(xiàn)其具有抑制乳腺腫瘤EMT6細(xì)胞的作用。但是蜂房的質(zhì)量標(biāo)準(zhǔn)方面,如顯微鑒別及薄層鑒別的文獻(xiàn)報道甚少,缺乏可靠的參考依據(jù),并且2020年版《中國藥典》也僅收載了蜂房的性狀鑒別,未收載顯微、薄層鑒別以及含量測定方法。因此,本實(shí)驗(yàn)首次對蜂房藥材的粉末特征和薄層色譜進(jìn)行觀察,并對其浸出物含量和沒食子酸含量進(jìn)行測定,測定方法快速簡便,重現(xiàn)性好,并建立了蜂房的質(zhì)量標(biāo)準(zhǔn),旨在提高蜂房藥材的質(zhì)量控制水平,為蜂房質(zhì)量標(biāo)準(zhǔn)的制定提供參考依據(jù)。
1 儀器與試藥
1.1 儀器
LC-10A高效液相色譜儀(日本島津公司),GH0525046C18A色譜柱(250×4.6mm,5μm,北京綠百草科技發(fā)展有限公司)。
1.2 試藥
蜂房(內(nèi)蒙古醫(yī)科大學(xué)附屬醫(yī)院中藥房,批號:121265-200301,由內(nèi)蒙古醫(yī)科大學(xué)藥學(xué)院李驍副教授鑒定),薄層層析硅膠(青島海洋化工有限公司,0110428)、羧甲基纖維素鈉(天津威晨化學(xué)試劑科貿(mào)有限公司,20070323)、沒食子酸標(biāo)準(zhǔn)品(中國藥品生物制品檢定所,生產(chǎn)批號110831-200302),蜂房對照藥材(中國藥品生物制品檢定所,批號:121265-200301),三氯甲烷、乙酸乙酯、甲酸、甲醇、無水乙醇等均為分析純。
2 方法與結(jié)果
2.1 蜂房的顯微特征
蜂房藥材粉末為灰褐色。光學(xué)顯微鏡下觀察可見蜂的剛毛淺黃色或黃色,散在且碎斷,長30~100 μm或更長,有的先端細(xì)尖或呈V型,有的前端細(xì)尖,基部突然膨大呈囊狀,有腔;蜂體壁碎片褐色或淺棕黃色,大小不等,分布散在,部分可見網(wǎng)狀紋理,少量呈魚鱗狀,碎片偶見瘤狀突起;蜂的橫紋肌淺黃色,具有紋理,平直,暗帶較窄;纖維呈絲狀,淺黃色,單個存在,部分成束;透明條狀物無色,彎曲處有褶皺;植物細(xì)胞偶見(見圖1)。
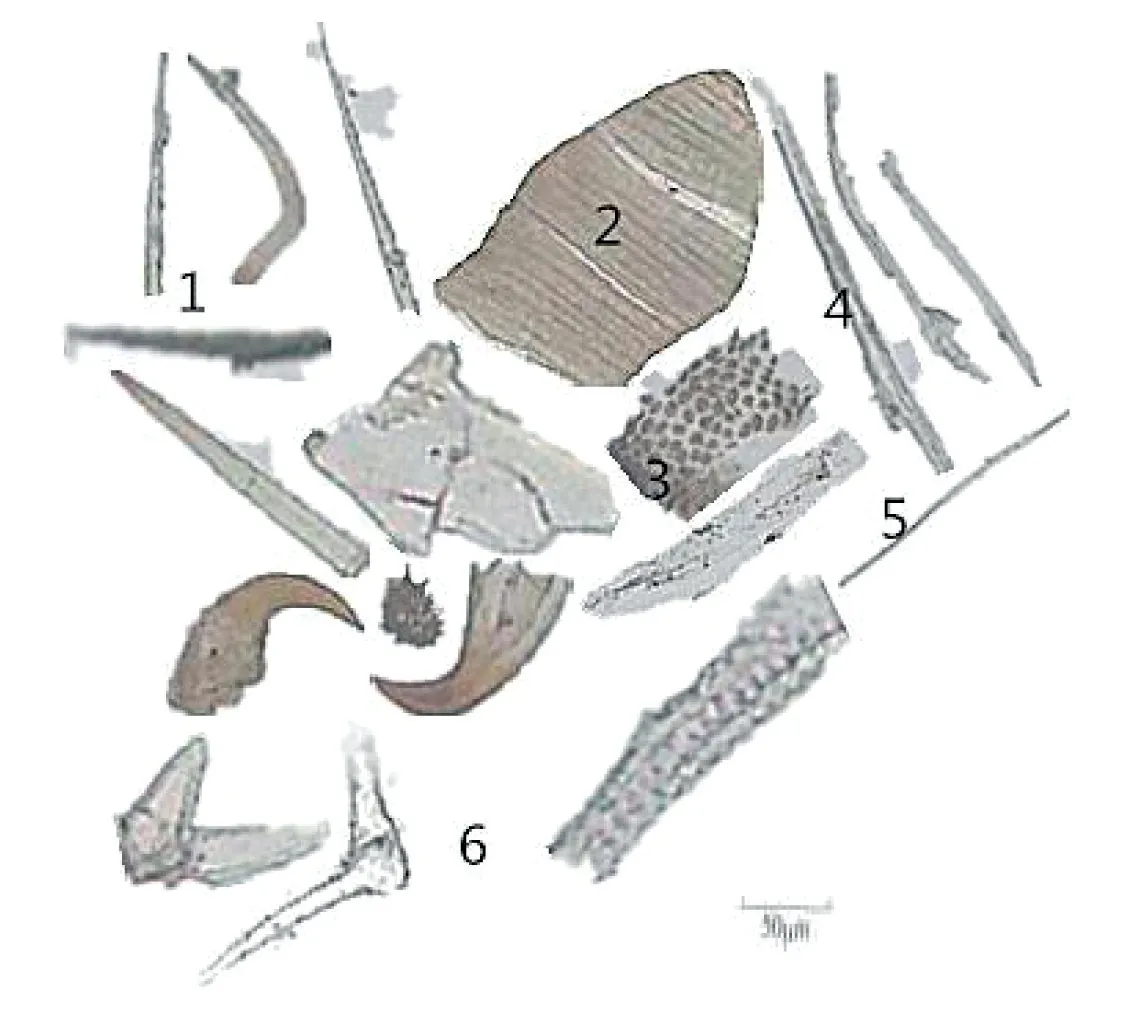
圖1 蜂房粉末顯微鑒別圖
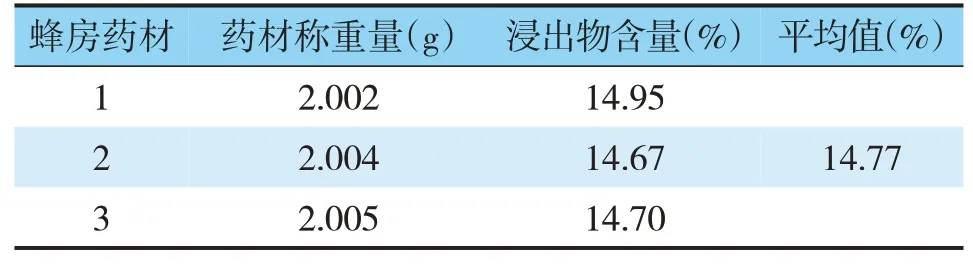
表2 蜂房熱浸法水浸出物含量
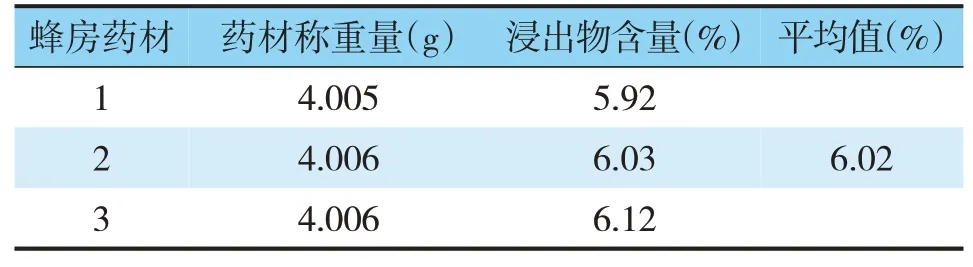
表3 蜂房冷浸法醇浸出物含量
2.2 蜂房的薄層色譜鑒別
2.2.1溶液制備[7]取蜂房藥材粉末3.5 g,20倍體積水加熱回流提取,濾液醇沉,濃縮上清液,加正丁醇萃取。回收有機(jī)相,甲醇溶解殘渣作為供試品溶液。取蜂房對照藥材3.5 g,同法制備對照藥材溶液。取標(biāo)準(zhǔn)品約5.10 mg,精密稱定,制得濃度為1.02 mg·mL-1的對照品甲醇溶液。
2.2.2薄層色譜分析 吸取蜂房供試品溶液、蜂房對照藥材溶液和對照品溶液各約4~6 μL,分別點(diǎn)于同一硅膠G薄層板。三氯甲烷-乙酸乙酯-甲酸(5:4:1)展開,取出,晾干,254 nm紫外燈下檢視(見圖2),樣品分離度良好。

圖2 蜂房粉末顯微鑒別圖
2.3 蜂房的浸出物檢查
取供試品,按照水溶性浸出物測定法和醇溶性浸出物測定法(《中國藥典》2015年版四部通則2201),分別采用水及乙醇作為浸出溶劑,冷、熱兩種浸出方法比較,測定結(jié)果(見表1~3)。結(jié)果顯示,以水為浸出溶劑熱浸法下,蜂房藥材浸出物的平均值為14.77%,浸出效率較高。

表1 蜂房冷浸法水浸出物含量
2.4 蜂房的含量測定
2.4.1對照品溶液的制備 取標(biāo)準(zhǔn)品1.32 mg,精密稱定,制得濃度為0.264 mg·mL-1的標(biāo)準(zhǔn)品甲醇溶液。
2.4.2供試品溶液的制備[8]取蜂房藥材粉末約2.0 g,精密稱定,加入100 mL的50%甲醇溶液,加熱回流提取1 h,放冷,過濾,濾液揮干,加甲醇定容于10 mL容量瓶,0.45 μm微孔濾膜濾過,即得。
2.4.3色譜條件依據(jù)查閱文獻(xiàn)及考察結(jié)果,確定色譜條件如下。采用北京綠百草科技發(fā)展有限公司GH0525046 C18A色譜柱(250×4.6 mm,5 μm),柱溫室溫,以甲醇-磷酸二氫鈉緩沖液(pH=3.0)(5:95)為流動相,梯度洗脫,流速1.0 mL·min-1,檢測波長273 nm,進(jìn)樣量10 μL。色譜圖(見圖3、4)。

圖3 沒食子酸標(biāo)準(zhǔn)品溶液高效液相色譜圖

圖4 蜂房供試品溶液高效液相色譜圖
2.4.4線性關(guān)系考察精密吸取“2.4.1”項下標(biāo)準(zhǔn)品溶 液0.1 mL、0.3 mL、0.5 mL、0.7 mL、0.9 mL,按“2.4.3”項下色譜條件進(jìn)樣。峰面積為X軸,標(biāo)準(zhǔn)品濃度為Y軸,得到標(biāo)準(zhǔn)曲線(見圖5)。沒食子酸線性考察結(jié)果表明,在5.28~47.52 μg·mL-1(R2=0.9993)濃度范圍內(nèi),其峰面積與濃度的線性關(guān)系良好。

圖5 沒食子酸標(biāo)準(zhǔn)曲線圖
2.4.5精密度試驗(yàn)精密吸取標(biāo)準(zhǔn)品溶液10 μL,按“2.4.3”項下色譜條件連續(xù)進(jìn)樣6次。記錄沒食子酸的色譜峰面積。結(jié)果顯示,沒食子酸峰面積RSD(n=6)為0.33%,精密度良好。
2.4.6穩(wěn)定性試驗(yàn)精密吸取供試品溶液10 μL,按“2.4.3”項下色譜條件在0、2、4、8、12、24 h分別進(jìn)樣6次,測定峰面積RSD(n=6)為1.58%。表明供試品溶液在24 h內(nèi)穩(wěn)定。
2.4.7重復(fù)性試驗(yàn)取同一批蜂房藥材6份,精密稱定,按“2.4.2”項下方法制備供試品溶液,按“2.4.3”項下色譜條件進(jìn)樣,測得沒食子酸平均含量為1.25 mg·g-1,RSD為1.85%。
2.4.8加樣回收率試驗(yàn)稱取已知含量的“2.4.7”項下蜂房藥材6份,每份約1.0 g,精密稱定。每份分別精密加入沒食子酸標(biāo)準(zhǔn)品3 mL、4 mL、5 mL、6 mL、7 mL、8 mL,按“2.4.2”項下方法制成樣品溶液,在“2.4.3”項下色譜條件進(jìn)樣,記錄色譜峰的面積。平均回收率測定結(jié)果(見表4)。

表4 加樣回收率試驗(yàn)結(jié)果(n=6)
2.4.9蜂房樣品的含量測定取蜂房藥材粉末三份,每份約1.0g,精密稱定,按“2.4.2”項下方法制備成蜂房樣品溶液,在“2.4.3”項下色譜條件進(jìn)樣,測定峰面積,計算蜂房樣品液中沒食子酸含量(見表5)。

表5 蜂房樣品含量測定結(jié)果(n=3)
3 討論
3.1 流動相的選擇[9~13]
本實(shí)驗(yàn)分別考察五種流動相體系(見表6)。分別按照上述條件等度洗脫后,結(jié)果顯示,pH值為3,配比為5:95的甲醇-磷酸二氫鈉緩沖液作為流動相時色譜峰分離情況較好,故選該體系為流動相。

表6 流動相條件表
3.2 紫外檢測波長的選擇
按照《中國藥典》2015年版三部制劑通則0401制備沒食子酸標(biāo)準(zhǔn)品稀釋液,于200-800 nm波長范圍內(nèi)進(jìn)行全波長光譜掃描,在273 nm附近沒食子酸吸光系數(shù)最大且干擾較少,故選擇273 nm作為吸收波長。
3.3 色譜條件的選擇
色譜條件優(yōu)化時,查閱相關(guān)文獻(xiàn),水相中多用的純水相或加有磷酸或乙酸。本實(shí)驗(yàn)發(fā)現(xiàn),水相中加入磷酸二氫鈉且將PH值調(diào)為酸性,分離效果明顯,且基線穩(wěn)定。實(shí)驗(yàn)中出現(xiàn)等度洗脫分離時間較長,雜質(zhì)峰分離度較差現(xiàn)象,因此進(jìn)行梯度洗脫。梯度洗脫時,混合對照品能很好的分離,但因?yàn)槌跏紳舛炔煌龇逦恢貌煌蛛x效果不同,經(jīng)過多次實(shí)驗(yàn)對比篩選,最終選定以35%有機(jī)相開始進(jìn)行梯度洗脫。
3.4 小結(jié)
本試驗(yàn)對蜂房的顯微、薄層特征進(jìn)行鑒別,對其浸出物及沒食子酸含量進(jìn)行了測定,以期為蜂房的質(zhì)量控制以及質(zhì)量標(biāo)準(zhǔn)的完善提供些許幫助。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(shù)(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質(zhì)量與標(biāo)準(zhǔn)化(2015年9期)2015-12-31 11:41:40
中國質(zhì)量與標(biāo)準(zhǔn)導(dǎo)報(2014年4期)2014-03-11 19:54:25
中國質(zhì)量與標(biāo)準(zhǔn)導(dǎo)報(2014年10期)2014-02-28 22:25:47
中國質(zhì)量與標(biāo)準(zhǔn)導(dǎo)報(2014年7期)2014-02-28 22:24:39
