QuEChERS結(jié)合UHPLC-MS/MS法測(cè)定畜肉中阿苯達(dá)唑及其代謝物的殘留量
2021-10-21 13:24:24黃永橋楊昌彪馬凱毛敏霞簡(jiǎn)銀池
食品與發(fā)酵工業(yè) 2021年19期
黃永橋,楊昌彪,馬凱*,毛敏霞,簡(jiǎn)銀池
1(貴州省檢測(cè)技術(shù)研究應(yīng)用中心,貴州 貴陽(yáng),550016) 2(貴州省分析測(cè)試研究院,貴州 貴陽(yáng),550016)
阿苯達(dá)唑又名丙硫咪唑,是苯并咪唑類驅(qū)蟲(chóng)藥物,因其治療效果好、毒性小等優(yōu)勢(shì),被廣泛用于治療動(dòng)物的腸道寄生蟲(chóng)感染病,是目前獸醫(yī)臨床使用最廣泛的藥物之一[1-2]。阿苯達(dá)唑進(jìn)入動(dòng)物體內(nèi)后會(huì)快速代謝為阿苯達(dá)唑亞砜,阿苯達(dá)唑亞砜進(jìn)一步轉(zhuǎn)化為阿苯達(dá)唑砜和阿苯達(dá)唑-2-氨基砜[3-4]。目前,對(duì)養(yǎng)殖中使用阿苯達(dá)唑藥物的管理日益加強(qiáng),對(duì)其及其代謝物進(jìn)行風(fēng)險(xiǎn)評(píng)估也極為重視,是動(dòng)物源性食品中獸藥殘留重點(diǎn)監(jiān)控對(duì)象之一,GB 31650—2019《食品安全國(guó)家標(biāo)準(zhǔn) 食品中獸藥最大殘留限量》規(guī)定了其在動(dòng)物性食品中的最大殘留量[5],動(dòng)物肌肉中最大殘留限量為100 μg/kg。
阿苯達(dá)唑及其代謝物常用檢測(cè)方法有高效液相色譜法(HPLC)[6-9],高效液相色譜-串聯(lián)質(zhì)譜法(HPLC-MS/MS)[10-13],超高效液相色譜-飛行時(shí)間質(zhì)譜聯(lián)用[14],氣相色譜-串聯(lián)質(zhì)譜法(GC-MS/MS)[15]等。其中HPLC-MS/MS法因其準(zhǔn)確度和靈敏度高、選擇性強(qiáng)等優(yōu)點(diǎn),廣泛用于動(dòng)物源性食品中獸藥殘留的分析測(cè)定。QuEChERS凈化方法前處理簡(jiǎn)單快速、溶劑使用量少,在農(nóng)獸藥殘留分析中應(yīng)用廣泛[16-19]。本研究擬利用QuEChERS技術(shù)結(jié)合超高效液相色譜-串聯(lián)質(zhì)譜法(ultra high performance liquid chromatography-tandem mass-spectrometry, UHPLC-MS/MS)分析技術(shù),通過(guò)優(yōu)化色譜和質(zhì)譜參數(shù)、提取溶劑和凈化方法等條件,建立了畜肉(豬肉、牛肉)中阿苯達(dá)唑及其代謝物殘留量的快速分析方法。本方法具有簡(jiǎn)單、快速、準(zhǔn)確和靈敏度高等優(yōu)點(diǎn),適用于畜肉(豬肉、牛肉)中阿苯達(dá)唑及其代謝物殘留量的快速篩查和定量分析,具有較強(qiáng)的實(shí)用價(jià)值。
1 材料與方法
1.1 儀器和試劑
1.1.1 主要儀器
Agilent 6470三重四級(jí)桿串聯(lián)質(zhì)譜儀,配有ESI源及Agilent 1290超高效液相色譜,美國(guó)Agilent公司;Blixer3攪拌機(jī),法國(guó)ROBOT COUPA公司;UMV-2多管渦旋混合器,北京普立泰科儀器有限公司;XW-80A渦旋混合器,上海米青科實(shí)業(yè)有限公司;陶瓷均質(zhì)子,迪馬科技有限公司;十八烷基鍵合硅膠吸附劑(C18)、乙二胺-N-丙基硅烷(primary secondary amine,PSA)、0.22 μm聚四氟乙烯(poly tetra fluoroethylene,PTFE)濾膜,上海安普實(shí)驗(yàn)科技股份有限公司。
1.1.2 主要試劑
甲醇、乙腈(色譜純),德國(guó)Merck公司;乙酸乙酯(分析純)、無(wú)水硫酸鎂,天津市科密歐化學(xué)試劑有限公司;甲酸(色譜純),上海安普實(shí)驗(yàn)科技股份有限公司;乙酸銨(分析純),西亞試劑;阿苯達(dá)唑、阿苯達(dá)唑砜、阿苯達(dá)唑亞砜、阿苯達(dá)唑-2-氨基砜(純度>98%),百靈威科技有限公司。
1.2 標(biāo)準(zhǔn)溶液配制
精密稱取阿苯達(dá)唑、阿苯達(dá)唑砜、阿苯達(dá)唑亞砜、阿苯達(dá)唑-2-氨基砜10 mg(精確至0.000 1 g)于100 mL棕色容量瓶中,用甲醇溶解并定容至刻度線,混勻,分別配制成質(zhì)量濃度為100 mg/L標(biāo)準(zhǔn)儲(chǔ)備液,-18 ℃避光保存。用甲醇將4種標(biāo)準(zhǔn)儲(chǔ)備液分別逐級(jí)稀釋至所需的混合標(biāo)準(zhǔn)工作液,現(xiàn)配現(xiàn)用。
1.3 色譜條件
色譜柱:ZORBAX Eclipse Plus-C18反相色譜柱(100 mm×2.1 mm,1.8 μm),美國(guó)Agilent公司;流動(dòng)相A:0.1%(體積分?jǐn)?shù))甲酸水溶液,B:乙腈;梯度洗脫程序:0.0~0.5 min,10% B;0.5~2.5 min,10%~90% B;2.5~3.0 min,90% B;3.0~3.1 min,90%~10% B;3.1~4.5 min,10%。流速:0.3 mL/min;柱溫40 ℃;進(jìn)樣體積5.0 μL。
1.4 質(zhì)譜條件
離子源:電噴霧離子源,正離子掃描;多反應(yīng)監(jiān)測(cè);毛細(xì)管電壓4.0 kV;霧化器壓力40 psi;干燥氣流速10 L/min;干燥氣溫度300 ℃;鞘氣流速10 L/min;鞘氣溫度300 ℃。
1.5 樣品前處理
稱取搗碎后的組織樣品5.00 g于50 mL具塞離心管中,準(zhǔn)確加入10 mL乙腈,加入1粒陶瓷均質(zhì)子,渦旋振蕩10 min,4 500 r/min常溫離心10 min,吸取2 mL上清液于15 mL離心管中,加入500 mg無(wú)水硫酸鎂和100 mg C18吸附劑,渦旋1 min,5 000 r/min離心2 min,取上清液過(guò)0.22 μm PTFE濾膜,待測(cè)。
1.6 基質(zhì)工作曲線配制
依次吸取不同體積的標(biāo)準(zhǔn)使用液加入相同取樣量的2種空白基質(zhì)樣品中(牛肉、豬肉),樣品按1.5前處理方法處理后得到2種基質(zhì)工作曲線,分別對(duì)2種樣品進(jìn)行定量校正。基質(zhì)標(biāo)準(zhǔn)工作曲線范圍分別為:阿苯達(dá)唑0.05~5.00 ng/mL,阿苯達(dá)唑砜0.10~10.00 ng/mL,阿苯達(dá)唑亞砜和阿苯達(dá)唑-2-氨基砜0.50~50.00 ng/mL。
2 結(jié)果與分析
2.1 樣品前處理的優(yōu)化
2.1.1 提取
選用基質(zhì)豬肉、牛肉陰性樣品,實(shí)驗(yàn)分別考察了乙腈、含0.1%(體積分?jǐn)?shù))甲酸的乙腈溶液、乙酸乙酯和含0.1%(體積分?jǐn)?shù))甲酸的乙酸乙酯溶液的提取效率。結(jié)果見(jiàn)圖1-a,結(jié)果表明,2種畜肉基質(zhì)中乙腈和含0.1%甲酸的乙腈溶液的提取效果明顯優(yōu)于乙酸乙酯和含0.1%甲酸的乙酸乙酯溶液的提取效果,且乙腈的提取效果略優(yōu)于含0.1%甲酸的乙腈溶液,因此本實(shí)驗(yàn)選擇乙腈作為樣品提取溶劑。
實(shí)驗(yàn)對(duì)渦旋振蕩提取的時(shí)間(5、10、15 min)進(jìn)行了考察,結(jié)果發(fā)現(xiàn),在5~10 min時(shí),隨著時(shí)間的延長(zhǎng)目標(biāo)物提取效果明顯增大,10 min以后隨著時(shí)間的增加,目標(biāo)物提取效果基本趨于穩(wěn)定,如圖1-b所示。故實(shí)驗(yàn)選用渦旋振蕩10 min作為樣品提取的時(shí)間。
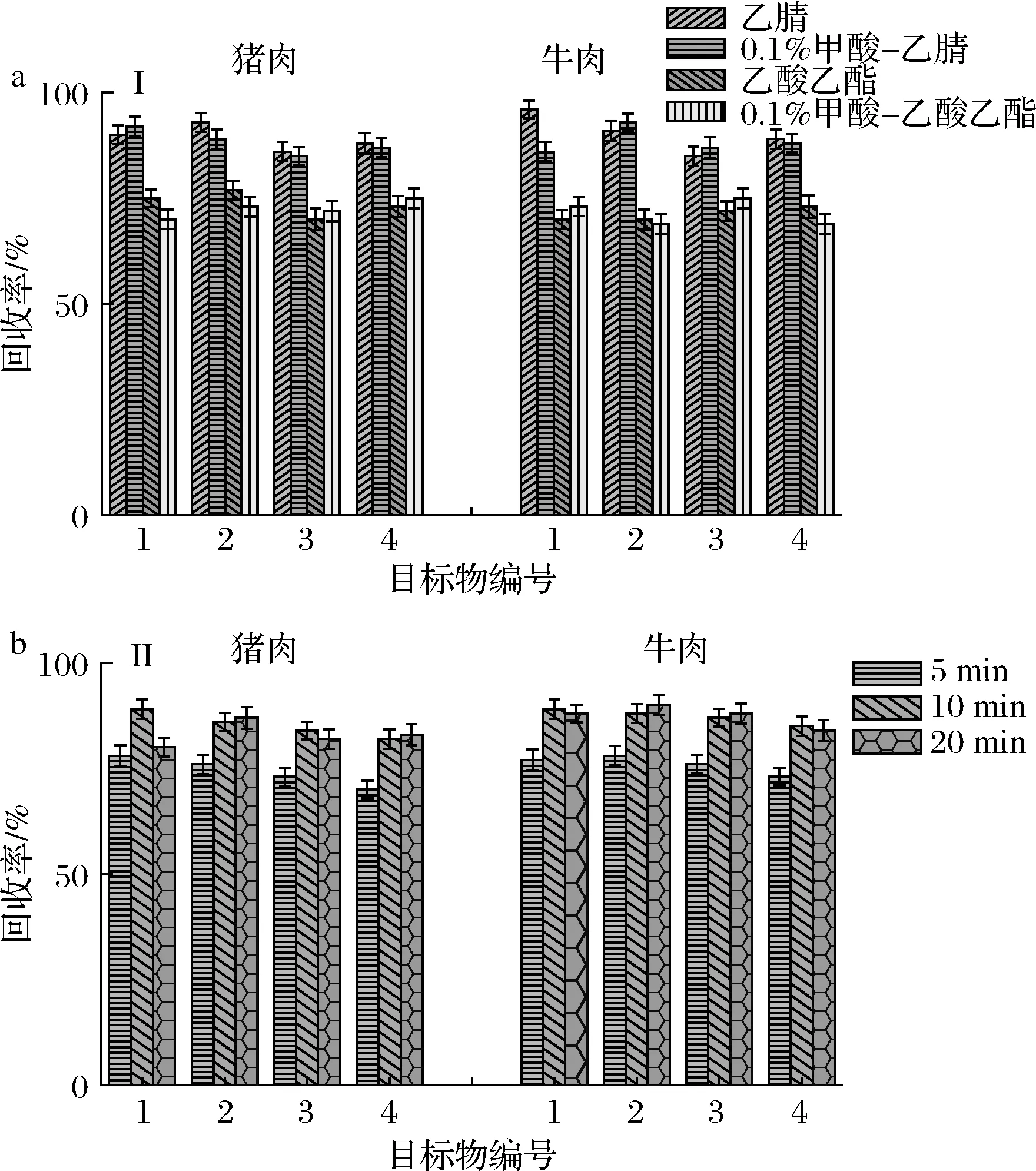
a-溶劑;b-時(shí)間1-阿苯達(dá)唑;2-阿苯達(dá)唑砜;3-阿苯達(dá)唑亞砜;4-阿苯達(dá)唑-2-氨基砜圖1 提取溶劑和提取時(shí)間對(duì)豬肉、牛肉中阿苯達(dá)唑及其代謝物的回收率范圍(n=3)Fig.1 Recovery rates of albendazole and its metabolites in pork and beef with extraction solvents and extraction times
2.1.2 凈化
目前,除了市售已有的產(chǎn)品,常用的吸附劑有C18、PSA等。本研究擬對(duì)QuEChERS dSPE EMR-Lipid凈化管、Oasis PRiME HLB固相萃取小柱(無(wú)須活化、上樣直接收集濾液)、PSA吸附劑及C18吸附劑凈化效果進(jìn)行了考察。結(jié)果如圖2-a,使用QuEChERS dSPE EMR-Lipid凈化管和Oasis PRiME HLB固相萃取小柱凈化時(shí),目標(biāo)物幾乎被填料完全吸附;使用PSA吸附劑時(shí)對(duì)目標(biāo)物有較強(qiáng)的吸附作用,導(dǎo)致目標(biāo)物損失較大;使用C18吸附劑凈化時(shí),目標(biāo)物損失較少,滿足相關(guān)要求。實(shí)驗(yàn)進(jìn)一步對(duì)C18吸附劑的用量(50、100、150 mg)進(jìn)行優(yōu)化。結(jié)果見(jiàn)圖2-b,使用100 mg C18吸附劑凈化樣品時(shí),凈化效果較好,且目標(biāo)物損失較少。最終本實(shí)驗(yàn)使用500 mg無(wú)水硫酸鎂和100 mg C18吸附劑作為樣品前處理方法。
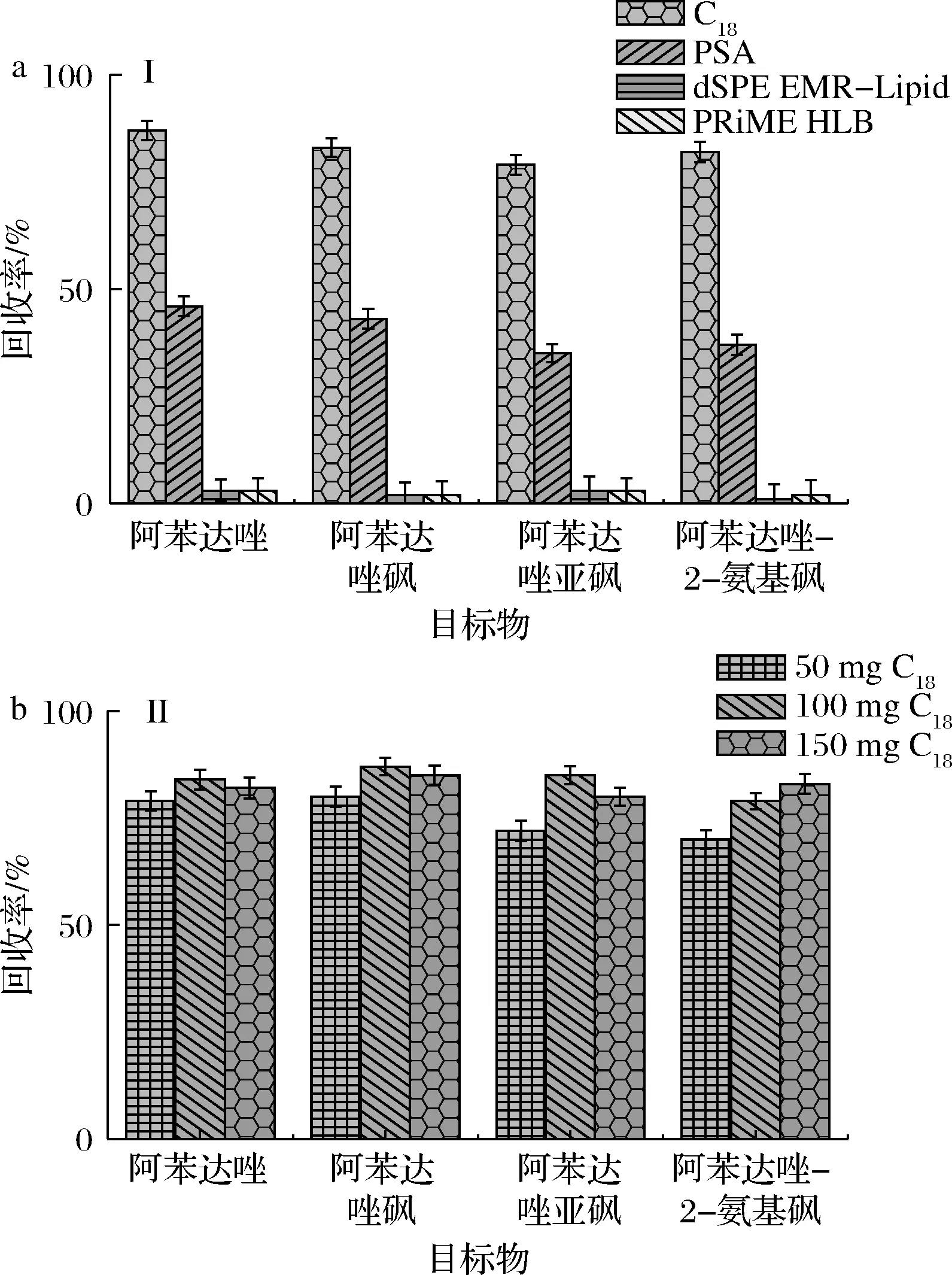
a-凈化試劑;b-C18用量圖2 凈化試劑及C18用量的優(yōu)化(n=3)Fig.2 Optimization of purification reagent and dosage of C18
2.2 色譜條件的優(yōu)化
本實(shí)驗(yàn)采用超高效液相色譜配合C18超高壓反相色譜柱對(duì)目標(biāo)物進(jìn)行分離。實(shí)驗(yàn)分別考察了乙腈-水、乙腈-0.1%甲酸水、乙腈-0.1%甲酸水(含2 mmol/L乙酸銨溶液)、甲醇-0.1%甲酸水、甲醇-0.1%甲酸水(含2 mmol/L乙酸銨溶液)等不同洗脫體系對(duì)目標(biāo)物分離能力。結(jié)果表明,使用乙腈-0.1%甲酸水作為流動(dòng)相時(shí),目標(biāo)物有良好的色譜分離效果,色譜峰峰型更好,且有較好的響應(yīng)值,進(jìn)一步對(duì)梯度洗脫程序進(jìn)行優(yōu)化,使目標(biāo)物有較好的分離,縮短樣品分析時(shí)間,提高了分析效率;實(shí)驗(yàn)考察了不同柱溫(25、30、40、50 ℃)對(duì)目標(biāo)物分離效果的影響,結(jié)果表明采用40 ℃的柱溫時(shí)目標(biāo)物有較好的分離效果。從而確立了最佳色譜條件,樣品單次分析時(shí)間僅需4.5 min,滿足大批樣品的快速檢測(cè)分析。
2.3 質(zhì)譜條件的優(yōu)化
配制100 μg/L的標(biāo)準(zhǔn)溶液采用不接色譜柱直接進(jìn)入質(zhì)譜的電噴霧離子源,在正離子模式下一級(jí)全掃描質(zhì)譜得到各目標(biāo)物分子離子峰([M+H]+),通過(guò)優(yōu)化去簇電壓,對(duì)母離子進(jìn)行二級(jí)質(zhì)譜分析,得到特征碎片離子質(zhì)譜圖,選擇特征碎片離子中響應(yīng)值高、基線噪聲低的離子對(duì)作為定性離子對(duì),選擇響應(yīng)值最高的離子對(duì)作為定量離子對(duì),優(yōu)化子離子對(duì)碰撞能量,使其豐度最大;進(jìn)一步優(yōu)化毛細(xì)管電壓、霧化器壓力、干燥和鞘氣的溫度及壓力等質(zhì)譜參數(shù),使其離子化效率最佳,得到最優(yōu)的質(zhì)譜條件,如表1。
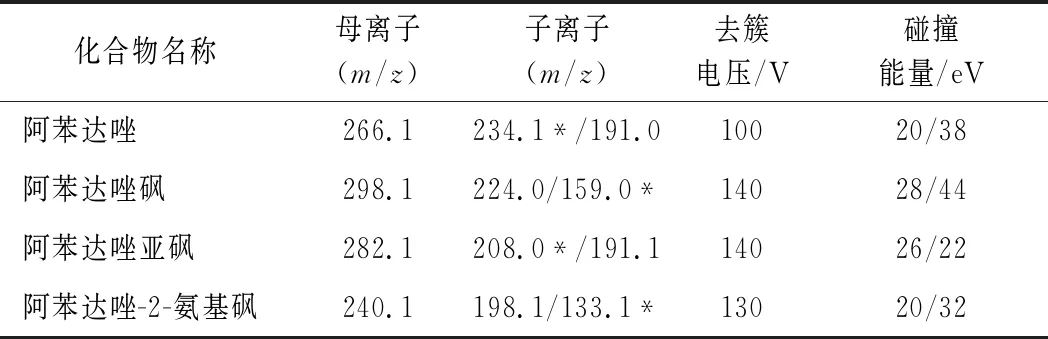
表1 質(zhì)譜參數(shù)Table 1 Mass spectrum conditions of albendazole and its metabolites
2.4 方法學(xué)評(píng)價(jià)
2.4.1 基質(zhì)效應(yīng)的評(píng)價(jià)及消除
基質(zhì)效應(yīng)(matrix effect,ME)是與目標(biāo)物共同洗脫并干擾質(zhì)譜儀電離過(guò)程的基質(zhì)物質(zhì)引起而導(dǎo)致目標(biāo)化合物信號(hào)強(qiáng)度有不同程度的增強(qiáng)或減弱的現(xiàn)象,包括基質(zhì)增強(qiáng)效應(yīng)和基質(zhì)抑制效應(yīng)[20],易影響儀器的靈敏度和分析結(jié)果的準(zhǔn)確性。因而在建立UHPLC-MS/MS檢測(cè)方法時(shí)應(yīng)對(duì)基質(zhì)效應(yīng)進(jìn)行評(píng)價(jià),為保證結(jié)果的準(zhǔn)確可靠,采取措施消除或減弱其影響是非常必要的。本方法采用空白樣品處理液匹配標(biāo)準(zhǔn)曲線和純?nèi)軇┢ヅ錁?biāo)準(zhǔn)曲線評(píng)價(jià)基質(zhì)效應(yīng),即ME=(基質(zhì)標(biāo)準(zhǔn)溶液曲線斜率/無(wú)基質(zhì)標(biāo)準(zhǔn)溶液曲線斜率-1)×100%,負(fù)值表示存在基質(zhì)抑制效應(yīng),正值表示存在基質(zhì)增強(qiáng)效應(yīng),絕對(duì)值越大則基質(zhì)效應(yīng)越強(qiáng)。本文在阿苯達(dá)唑及其代謝物標(biāo)準(zhǔn)曲線范圍濃度內(nèi)進(jìn)行評(píng)價(jià)(表2),可以看出阿苯達(dá)唑及其代謝物均存在較強(qiáng)的基質(zhì)效應(yīng),阿苯達(dá)唑和阿苯達(dá)唑-2-氨基砜為基質(zhì)抑制效應(yīng),阿苯達(dá)唑砜和阿苯達(dá)唑亞砜為基質(zhì)增強(qiáng)效應(yīng)。
2.4.2 方法的標(biāo)準(zhǔn)曲線和檢出限
本方法采用空白樣液稀釋混合標(biāo)準(zhǔn)工作曲線,制備成系列不同濃度的混合標(biāo)準(zhǔn)工作液,在選定的色譜和質(zhì)譜條件下測(cè)定,以標(biāo)準(zhǔn)溶液定量離子對(duì)峰面積(y)對(duì)其質(zhì)量濃度(x)做標(biāo)準(zhǔn)曲線,得到阿苯達(dá)唑及其代謝物的工作曲線。結(jié)果表明,阿苯達(dá)唑及其代謝物各自的質(zhì)量濃度范圍內(nèi),呈現(xiàn)良好的線性關(guān)系,相關(guān)系數(shù)(R2)均>0.998,適用于定量分析。通過(guò)向陰性樣品中添加目標(biāo)物來(lái)考察方法的檢出限(S/N=3)和定量限(S/N=10),最終確定方法的檢出限為(μg/kg):阿苯達(dá)唑-2-氨基砜1 ;阿苯達(dá)唑亞砜1;阿苯達(dá)唑砜0.2;阿苯達(dá)唑0.1。定量限為(μg/kg):阿苯達(dá)唑-2-氨基砜3;阿苯達(dá)唑亞砜3;阿苯達(dá)唑砜0.6;阿苯達(dá)唑0.3。結(jié)果見(jiàn)表2,說(shuō)明該方法具有較好的靈敏度。
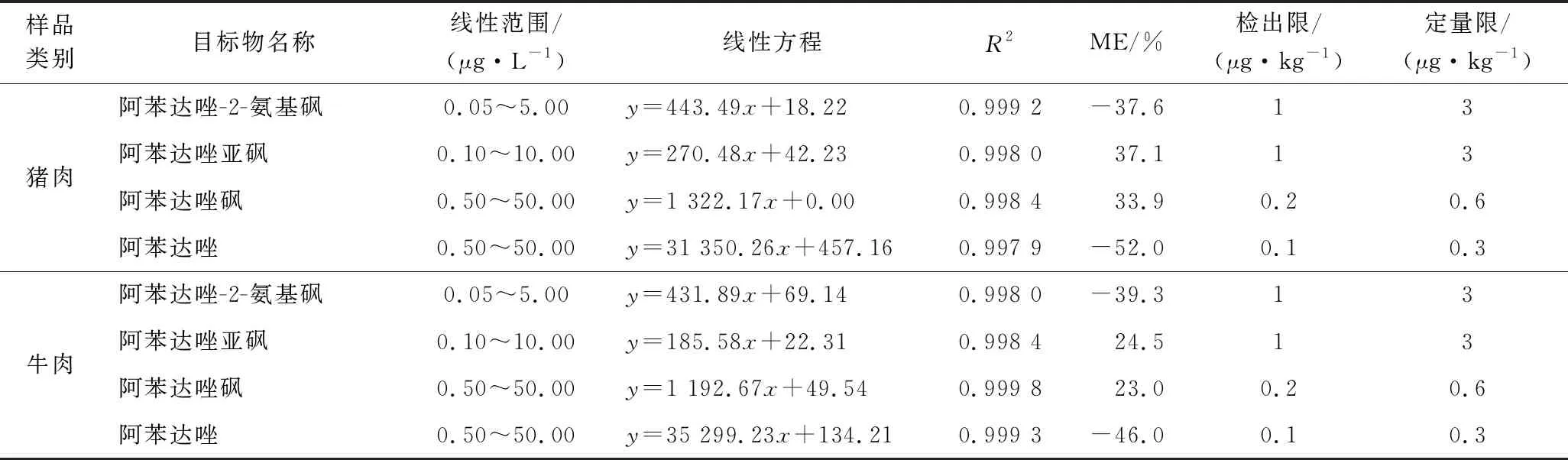
表2 方法線性方程、相關(guān)系數(shù)、基質(zhì)效應(yīng)、檢出限及定量限Table 2 Calibration equation,correlation coefficient,ME,LOD and LOQ of the method
2.5 加標(biāo)回收及精密度考察
選取豬肉和牛肉陰性樣品,分別添加檢出限的1、2、10倍3個(gè)不同濃度,每個(gè)添加水平做6個(gè)平行,按照1.5中的樣品前處理方法進(jìn)行提取凈化,3個(gè)加標(biāo)濃度連續(xù)做3 d,分別計(jì)算平均回收率和精密度,精密度以相對(duì)標(biāo)準(zhǔn)偏差(relative standard deviation,RSD)表示。結(jié)果如表3所示,3個(gè)添加水平的準(zhǔn)確度均<15%,日內(nèi)(n=6)平均回收率分別為豬肉:81.9%~109.8%,牛肉:82.3%~103.5%;日內(nèi)(n=6)RSD分別為豬肉:3.3%~12.5%,牛肉:1.8%~9.0%。日間(n=3)平均回收率分別為豬肉:81.8%~99.0%,牛肉:86.3%~99.0%;日間(n=3)RSD分別為豬肉:3.1%~12.6%,牛肉:2.4%~14.4%。說(shuō)明該方法具有較好的重復(fù)性與準(zhǔn)確性,能夠滿足豬肉和牛肉中阿苯達(dá)唑及其代謝物殘留量的測(cè)定。圖3為混合標(biāo)準(zhǔn)溶液、空白樣品和加標(biāo)樣品的總離子流圖。
2.6 實(shí)際樣品的測(cè)定
利用本研究建立的方法對(duì)市售的50批次豬肉樣品、38批次牛肉樣品進(jìn)行測(cè)定,其中1份豬肉樣品和1份牛肉樣品中阿苯達(dá)唑-2-氨基砜檢測(cè)結(jié)果為陽(yáng)性,含量分別為3.40、32.1 μg/kg,其余樣品均未檢出或低于定量限。
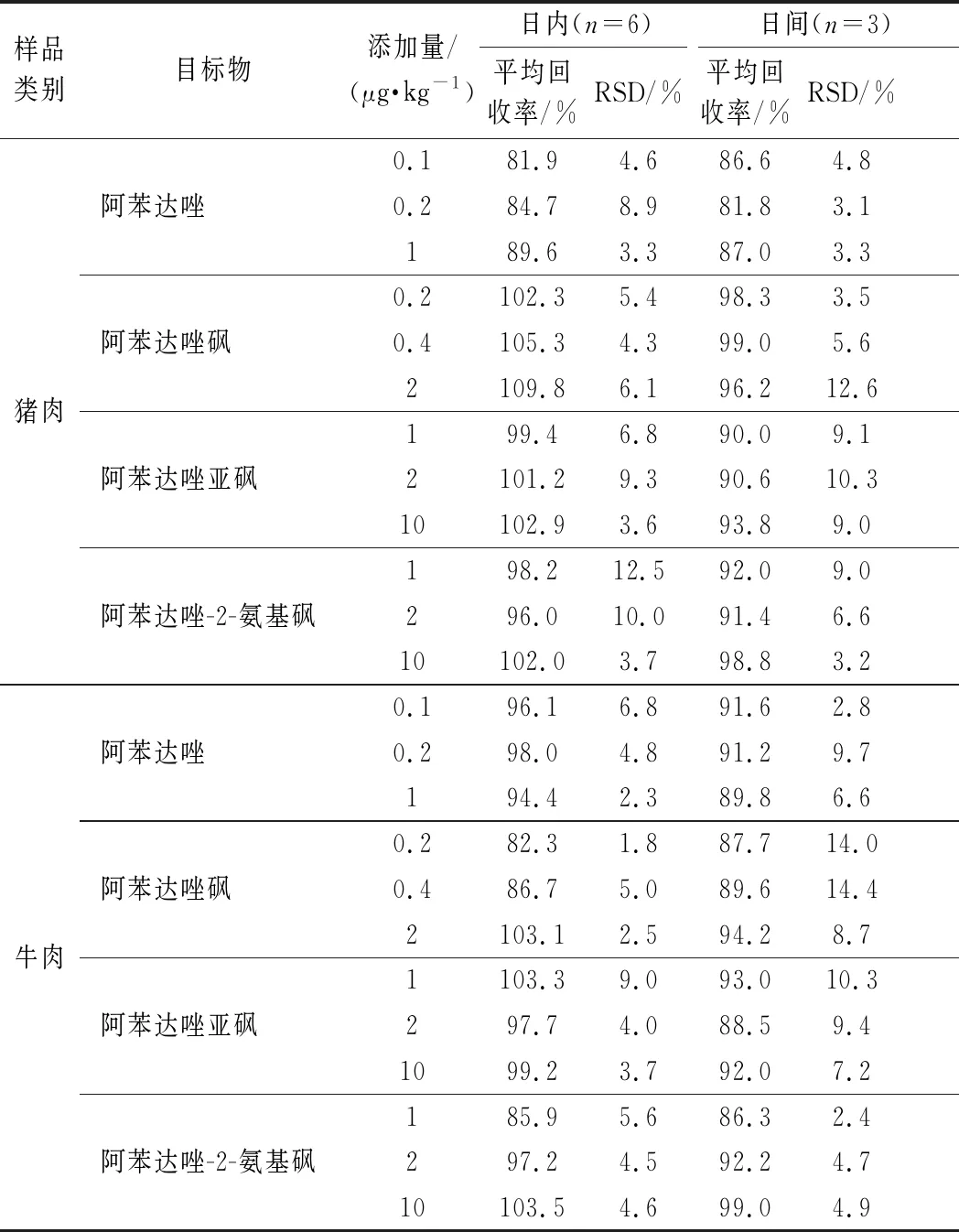
表3 回收率和精密度Table 3 Recoveries and precision of the method
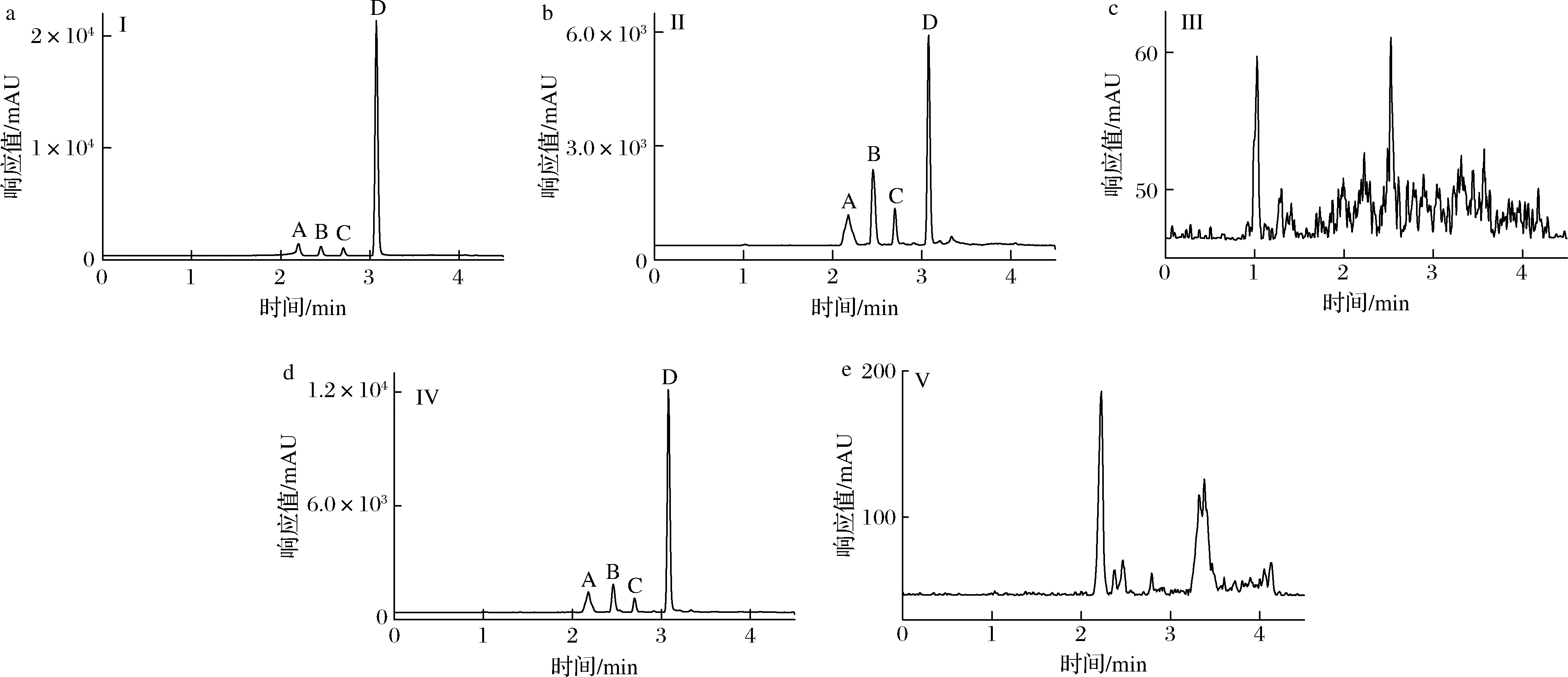
a-4種阿苯達(dá)唑混合標(biāo)準(zhǔn)溶液(阿苯達(dá)唑0.5 μg/L,阿苯達(dá)唑砜1 μg/L,阿苯達(dá)唑亞砜和阿苯達(dá)唑-2-氨基砜5 μg/L);b-豬肉樣品加標(biāo)(阿苯達(dá)唑1 μg/kg,阿苯達(dá)唑砜2 μg/kg,阿苯達(dá)唑亞砜和阿苯達(dá)唑-2-氨基砜10 μg/kg);c-牛肉樣品加標(biāo)(阿苯達(dá)唑1 μg/kg,阿苯達(dá)唑砜2 μg/kg,阿苯達(dá)唑亞砜和阿苯達(dá)唑-2-氨基砜10 μg/kg);d-豬肉空白樣品;e-牛肉空白樣品;A-阿苯達(dá)唑-2-氨基砜;B-阿苯達(dá)唑亞砜;C-阿苯達(dá)唑砜;D-阿苯達(dá)唑圖3 混合標(biāo)準(zhǔn)溶液、豬肉樣品加標(biāo)、豬肉、牛肉樣品加標(biāo)和牛肉總離子流圖Fig.3 Total ion chromatograms of standard solution,pork sample spiked with albendazole and its metabolites,pork blank sample,beef sample spiked with albendazole and its metabolites and beef blank sample
3 結(jié)論
本研究采用高效液相色譜-串聯(lián)質(zhì)譜儀建立了豬肉、牛肉中阿苯達(dá)唑及其代謝物(阿苯達(dá)唑砜、阿苯達(dá)唑亞砜、阿苯達(dá)唑-2-氨基砜)殘留量的快速分析方法,通過(guò)對(duì)樣品前處理、液相色譜、質(zhì)譜等條件進(jìn)行了優(yōu)化,并進(jìn)行了方法學(xué)考察。結(jié)果表明,阿苯達(dá)唑及其代謝物在4.5 min內(nèi)完成分析,且目標(biāo)物有較好的分離度,相關(guān)系數(shù)(R2)均>0.998,檢測(cè)限為0.1~1 μg/kg,定量限在0.3~3 μg/kg,日內(nèi)(n=6)平均回收率在81.9%~109.8%,日間(n=3)平均回收率在81.8%~99.0%,精密度為1.8%~14.4%。本方法操作簡(jiǎn)單、快速、準(zhǔn)確、靈敏度高,重現(xiàn)性好,回收率高且穩(wěn)定,檢測(cè)限及定量限低于現(xiàn)有檢測(cè)方法要求,能夠準(zhǔn)確定性和定量分析畜肉(豬肉、牛肉)中阿苯達(dá)唑及其代謝物殘留量,滿足日常分析檢測(cè)的要求。
