氧化亞油酸對(duì)肌原纖維蛋白膠凝行為及熱誘導(dǎo)凝膠體外消化率的影響
2021-10-22 00:21:58李保玲李穎朱振寶黃峻榕曹云剛
食品與發(fā)酵工業(yè) 2021年19期
李保玲,李穎,朱振寶,黃峻榕,曹云剛
(陜西科技大學(xué) 食品與生物工程學(xué)院,陜西 西安,710021)
肉及其制品的質(zhì)地特征是影響消費(fèi)者做出購(gòu)買(mǎi)決定的重要因素,占肌肉蛋白總量55%~65%的肌原纖維蛋白因?yàn)槠鋬?yōu)異的凝膠和乳化能力,對(duì)最終加工肉制品的質(zhì)地特性起著主要決定作用[1]。因此在肉制品加工過(guò)程中,蛋白結(jié)構(gòu)特征的變化及凝膠精細(xì)網(wǎng)絡(luò)的形成受到了廣泛關(guān)注[2-3]。在肉類食品加工過(guò)程中,肌肉細(xì)胞結(jié)構(gòu)的破壞、分子氧的滲透以及游離鐵和血紅素鐵的釋放不可避免地導(dǎo)致了脂肪和蛋白質(zhì)的氧化。長(zhǎng)期以來(lái)人們對(duì)脂質(zhì)的氧化進(jìn)行了深入的研究,然而蛋白質(zhì)作為活性氧(reactive oxygen species,ROS)或者其他過(guò)氧化物作用的靶點(diǎn)卻一直被忽視[4-5]。之前的研究主要聚焦于蛋白質(zhì)氧化在年齡相關(guān)疾病中的作用[6-7],但目前越來(lái)越多的研究開(kāi)始探討蛋白質(zhì)氧化對(duì)食品質(zhì)量的影響。ROS 被認(rèn)為是與氨基酸側(cè)鏈基團(tuán)的氧化修飾有關(guān)的原位因素,通常會(huì)導(dǎo)致蛋白質(zhì)結(jié)構(gòu)與功能性質(zhì)的改變,包括持水性能、凝膠性能以及乳化性能等[5,8]。
由于原料肉中蛋白質(zhì)和不飽和脂肪大量共存以及加工行為的破壞性作用,脂肪和蛋白的氧化極可能以相關(guān)聯(lián)的方式發(fā)生。相關(guān)研究表明,脂質(zhì)氫過(guò)氧化物、脂質(zhì)自由基以及衍生的醛類物質(zhì)等都可以促進(jìn)蛋白氧化的發(fā)生[9-10]。關(guān)于脂質(zhì)氧化引起的肌原纖維蛋白(myofibrillar protein,MP)生化指標(biāo)和蛋白消化率的變化已有大量研究報(bào)道[11-14],但是由脂質(zhì)氧化產(chǎn)物導(dǎo)致的MP凝膠性能改變的現(xiàn)有研究結(jié)果并不一致,同時(shí)前人研究多集中于氧化對(duì)生蛋白消化率的影響,而現(xiàn)實(shí)生活中人們對(duì)肉食的攝入主要以熟食為主。因此,本實(shí)驗(yàn)主要研究蛋白暴露在脂肪氧合酶——亞油酸氧化體系中所引起的MP結(jié)構(gòu)以及凝膠性能的變化和氧化對(duì)蛋白熱誘導(dǎo)凝膠體外消化率的影響,以此進(jìn)一步了解肉類及其制品中脂質(zhì)氧化和蛋白質(zhì)氧化之間的關(guān)系。
1 材料與方法
1.1 實(shí)驗(yàn)材料
豬外脊肉(Longissimuslumborum),當(dāng)?shù)爻?陜西西安);大豆脂肪氧合酶、胰蛋白酶,美國(guó)Sigma-Aldrich公司;亞油酸(>95%)、胃蛋白酶,上海阿拉丁化學(xué)有限公司;其他試劑至少為分析純。
1.2 儀器與設(shè)備
HR/T20MM立式高速冷凍離心機(jī),湖南赫西儀器裝備有限公司;TA.Plus物性測(cè)試儀,英國(guó)Stable Micro System公司;Fluoro Max-4 熒光分光光度計(jì),日本 Horiba 公司;Mastersizer 2000激光粒度分析儀,英國(guó)Malvern Instruments有限公司;CM-5分光測(cè)色計(jì),柯尼卡美能達(dá)(中國(guó))投資有限公司;Haake-Mars 60 流變儀、DXR2顯微共聚焦拉曼光譜儀,德國(guó)Thermo Fisher Scientific公司;FEI Q45+EDAX Octane Prime環(huán)境掃描電子顯微鏡,美國(guó)FEI公司;安東帕LitesizerTM500納米粒度及Zeta電位分析儀,奧地利Anton Paar 公司。
1.3 MP的提取及樣品制備
1.3.1 MP的提取
肌原纖維蛋白的提取參照PARK等[13]的方法,使用雙縮脲法以牛血清白蛋白(bovine serum albumin,BSA)作為標(biāo)準(zhǔn)測(cè)定蛋白濃度,將所得MP置于碎冰浴4 ℃保存并在48 h內(nèi)使用。
1.3.2 蛋白的氧化處理
用25 mmol/L的磷酸鹽緩沖液(含0.4 mol/L NaCl,pH 6.2)將MP稀釋至45 mg/mL,將MP溶液分散于脂肪氧化體系(含0.5~10.0 mmol/L 亞油酸,3 750 U/mL脂肪氧合酶,終濃度)使MP終濃度達(dá)到30 mg/mL,并于4 ℃反應(yīng)12 h。通過(guò)添加Trolox(終濃度 1 mmol/L)來(lái)終止氧化反應(yīng),未添加氧化體系的MP溶液為空白對(duì)照。
1.4 脂質(zhì)氧化的測(cè)定
參照SALIH等[15]的方法測(cè)定2-硫代巴比妥酸反應(yīng)物(thiobarbituric acid reactive substances,TBARS)含量,并以此來(lái)反映MP懸液中的脂質(zhì)氧化程度。
1.5 MP的化學(xué)及結(jié)構(gòu)變化
1.5.1 氨基酸殘基側(cè)鏈修飾
羰基含量采用CAO等[16]描述的2,4-二硝基苯肼(2,4-dinitrophenylhydrazine,DNPH)比色法進(jìn)行測(cè)定。
總巰基含量按照ELLMAN[17]的方法,采用5,5’-二硫代雙(2-硝基苯甲酸)[5,5'-dithiobis-(2-nitrobenzoic acid),DTNB]進(jìn)行測(cè)定。
自由氨基含量參照HABEEB[18]的方法,通過(guò)2,4,6-三硝基苯磺酸(2,4,6-trinitrobenzenesulfonic acid,TNBS)試劑進(jìn)行測(cè)定,同時(shí)使用一系列濃度的L-亮氨酸在相同條件下制成標(biāo)準(zhǔn)曲線,并通過(guò)繪制的標(biāo)準(zhǔn)曲線來(lái)計(jì)算蛋白樣品的自由氨基含量。
1.5.2 蛋白構(gòu)象變化
內(nèi)源性色氨酸熒光的測(cè)定:使用25 mmol/L的磷酸鹽緩沖液(含0.4 mol/L NaCl,pH 6.2)將樣品稀釋至0.4 mg/mL,利用熒光分光光度計(jì)于波長(zhǎng)283 nm激發(fā)并記錄290~400 nm的發(fā)射光譜,激發(fā)和發(fā)射狹縫寬度均設(shè)置為1.5 nm。相同條件下記錄溶劑發(fā)射光譜,并從樣品發(fā)射光譜中扣除以排除干擾。
1.6 Zeta電位和粒度測(cè)定
Zeta電位:利用電位分析儀進(jìn)行測(cè)定,使用25 mmol/L磷酸鹽緩沖液(含0.4 mol/L NaCl,pH 6.2)將樣品稀釋至1 mg/mL,取1 mL放入U(xiǎn)nivette樣品池,平衡時(shí)間設(shè)定為1 min,設(shè)置溶劑(磷酸鹽緩沖液)折射率為1.331 8,電泳溫度維持在25 ℃,梯度重復(fù)3次,取其平均值。
粒度測(cè)定:使用激光粒度分析儀在25 ℃下采用靜態(tài)光散射分析MP樣品的粒徑分布,去除測(cè)量背景后將稀釋后的MP樣品(2 mg/mL)分散在蒸餾水(分散介質(zhì))中,直至遮光效果達(dá)到10%~13%,設(shè)置MP顆粒折射率為1.434,分散劑折射率為1.330,測(cè)定樣品的平均粒度(d3,2和d4,3)。
1.7 溶解度
使用25 mmol/L的磷酸鹽緩沖液(含0.4 mol/L NaCl,pH 6.2)將肌原纖維蛋白樣品稀釋至 2 mg/mL,然后在5 000×g條件下離心(15 min,4 ℃)。取上清液根據(jù)CAO等[1]的方法測(cè)定得到的蛋白樣品的溶解度。
1.8 十二烷基硫酸鈉聚丙烯酰胺凝膠電泳(polyacrylamide gel electrophoresis,SDS-PAGE)
采用SDS-PAGE分析了蛋白質(zhì)組分的氧化交聯(lián)、聚合或降解情況。實(shí)驗(yàn)參照CAO等[19]的方法進(jìn)行,使用4%濃縮膠和12%分離膠在還原(+DTT)和非還原(-DTT)條件下進(jìn)行凝膠電泳,每孔上樣量20 μg,染色脫色后拍照并對(duì)電泳條帶進(jìn)行分析。
1.9 凝膠性能測(cè)定
1.9.1 動(dòng)態(tài)流變性能測(cè)定
將不同處理的MP樣品離心脫氣后(30 mg/mL,1 000×g,1 min),取適量樣品均勻涂布于流變儀的平板上,選用平行板傳感器P35(直徑35 mm)下壓至1 mm處,用紙去除周圍多余樣品,在邊緣涂上硅油防止水分蒸發(fā),在振蕩模式下從 20 ℃加熱到 75 ℃,升溫速率:1 ℃/min,振蕩頻率:0.1 Hz,控制最大應(yīng)變:0.02。凝膠性能通過(guò)儲(chǔ)存模量(G′)來(lái)進(jìn)行評(píng)價(jià)。
1.9.2 蒸煮損失、凝膠強(qiáng)度和白度
將MP樣品(5 g,30 mg/mL)置于小玻璃瓶中使用保鮮膜密封后置于水浴鍋中,以1 ℃/min的升溫速率從20 ℃加熱至75 ℃,并在75 ℃保溫10 min,取出后立刻置于冰水混合物中冷卻30 min,隨后放入4 ℃冰箱冷藏過(guò)夜。蒸煮損失、凝膠強(qiáng)度和白度的測(cè)定參照CAO等[1]的方法進(jìn)行。
1.9.3 MP凝膠微觀結(jié)構(gòu)
將制備的MP凝膠切成小塊(5 mm×5 mm×5 mm),用含2.5%(體積分?jǐn)?shù))戊二醛的磷酸鹽緩沖液(pH 7.4)固定4 h,使用磷酸鹽緩沖液清洗3次,然后通過(guò)一系列濃度的乙醇溶液(50%,70%,90%,95%,100%體積分?jǐn)?shù))進(jìn)行梯度脫水,每次30 min。在-70 ℃對(duì)樣品進(jìn)行冷凍干燥,將干燥的樣品噴金后使用環(huán)境掃描電子顯微鏡(environmental scanning electron microscope,ESEM)觀察其微觀結(jié)構(gòu),加速電壓15 kV,放大倍數(shù)3 500倍。
1.9.4 MP凝膠的疏水相互作用和氫鍵作用
采用拉曼光譜對(duì)MP凝膠中760 cm-1附近的拉曼譜帶歸一化強(qiáng)度和830、850 cm-1處酪氨酸雙譜帶的相對(duì)強(qiáng)度進(jìn)行了表征[20],探究氧化對(duì)MP凝膠疏水相互作用和氫鍵作用的影響,光譜數(shù)據(jù)由OMNIC軟件收集后使用LabSpec對(duì)所得光譜進(jìn)行平滑、基線校正并用在1 003 cm-1處的苯丙氨酸環(huán)帶進(jìn)行歸一化處理[21]。
1.9.5 MP凝膠的體外消化
參照曹云剛[22]的方法模擬胃-腸道消化對(duì)MP凝膠進(jìn)行體外消化評(píng)價(jià)。取冷卻過(guò)夜的MP凝膠及其流出液5 g,磨碎后加入適量10 mmol/L的HCl溶液,在11 000 r/min條件下均質(zhì)30 s,添加豬胃蛋白酶溶液(1 mg/mL,溶于10 mmol/L HCl溶液),所得混合溶液蛋白質(zhì)量濃度為 3 mg/mL,胃蛋白酶濃度4%(質(zhì)量分?jǐn)?shù),以蛋白含量為基準(zhǔn)計(jì)算),pH 2.0。混勻后于37 ℃水浴鍋中酶解1 h,而后使用1 mol/L的NaOH溶液調(diào)節(jié)pH值至7.5來(lái)結(jié)束反應(yīng),而后添加4%(質(zhì)量分?jǐn)?shù),以蛋白含量為基準(zhǔn)計(jì)算)的胰酶溶液,繼續(xù)在37 ℃水浴鍋中反應(yīng)2 h,反應(yīng)結(jié)束后于沸水浴中加熱5 min以達(dá)到滅活的目的。消化反應(yīng)期間分別在30、60、90、120、180 min時(shí)取樣,加入同等體積的30% (質(zhì)量分?jǐn)?shù))三氯乙酸來(lái)終止反應(yīng)沉淀蛋白,混合液在4 ℃冰箱中過(guò)夜,以11 000×g離心10 min后棄去上清液,于沉淀中加入1 mol/L的NaOH溶液來(lái)復(fù)溶蛋白沉淀,隨后使用雙縮脲法測(cè)定蛋白濃度。體外消化率計(jì)算如公式(1)所示:
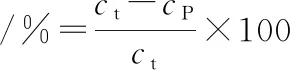
(1)
式中:ct和cp分別指總蛋白濃度以及體外消化后三氯乙酸沉淀的蛋白濃度。
1.10 數(shù)據(jù)分析
所有的實(shí)驗(yàn)至少獨(dú)立重復(fù)2次,每批次至少有3個(gè)獨(dú)立樣品。實(shí)驗(yàn)數(shù)據(jù)使用Statistix 9.0分析軟件進(jìn)行方差分析,采用LSD對(duì)多組數(shù)據(jù)進(jìn)行顯著性分析,P<0.05表示差異有統(tǒng)計(jì)學(xué)意義。數(shù)據(jù)以平均數(shù)±標(biāo)準(zhǔn)差(mean±SD)表示,并使用Sigma Plot 12.5軟件進(jìn)行繪圖。
2 結(jié)果與分析
2.1 脂質(zhì)氧化
相關(guān)研究發(fā)現(xiàn),多不飽和脂肪酸(亞油酸、亞麻酸等)在脂肪氧合酶的催化下發(fā)生脂質(zhì)過(guò)氧化反應(yīng),形成自由基中間體和相關(guān)脂質(zhì)氧化產(chǎn)物,進(jìn)而促進(jìn)MP氧化[12]。本研究采用TBARS法通過(guò)測(cè)定脂質(zhì)氧化二級(jí)產(chǎn)物的含量來(lái)評(píng)價(jià)脂質(zhì)氧化程度。如表1所示,未氧化MP的TBARS值為0.94 mg/kg蛋白,這與先前研究報(bào)告的值(0.79 mg/kg)相似[23]。總的來(lái)說(shuō),隨著OLA濃度的增加,脂質(zhì)氧化MP樣品TBARS值不斷增加,在OLA濃度為10.0 mmol/L時(shí),TBARS值達(dá)到最大(4.86 mg/kg),是對(duì)照樣品的5倍。這一結(jié)果說(shuō)明OLA濃度的增加促進(jìn)了脂質(zhì)氧化反應(yīng)的發(fā)生。
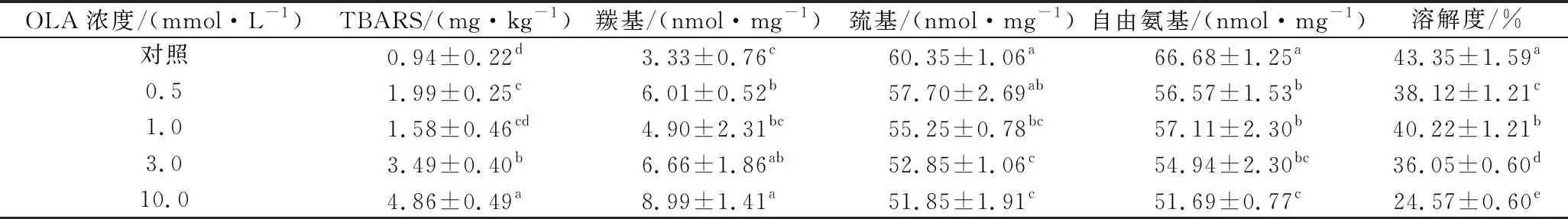
表1 不同OLA濃度下肌原纖維蛋白理化性質(zhì)的變化Table 1 Changes in physicochemical properties of myofibrillar protein at different concentrations of OLA
2.2 MP樣品氨基酸殘基的側(cè)鏈修飾情況
2.2.1 羰基
蛋白質(zhì)羰基含量的變化是反映蛋白質(zhì)氧化程度的重要指標(biāo),它主要由氨基酸側(cè)鏈殘基和肽鍵的氧化斷裂產(chǎn)生[5]。OLA濃度對(duì)MP羰基含量的影響如表1所示,未氧化MP的羰基含量為3.33 nmol/mg,這一結(jié)果與JONGBERG等[24]研究的未氧化豬肉MP的羰基值相接近。然而,也有部分研究報(bào)道的未氧化豬肉MP的羰基值較低[11,14],推測(cè)實(shí)驗(yàn)數(shù)據(jù)的不一致與實(shí)驗(yàn)所用豬肉的品種及屠宰方式有關(guān)。與TBARS含量變化趨勢(shì)一致,隨著OLA濃度的增加,脂質(zhì)氧化MP中羰基也在不斷生成,在OLA濃度為10.0 mmol/L時(shí)羰基含量達(dá)到最大值(8.99 nmol/mg,P<0.05),其他研究者也報(bào)道了脂質(zhì)氧化MP中羰基含量的類似變化趨勢(shì)[11,14]。這些結(jié)果表明脂質(zhì)氧化促進(jìn)了蛋白氧化的發(fā)生,同時(shí)也證實(shí)了脂質(zhì)氧化產(chǎn)物可以有效催化蛋白氧化的觀點(diǎn)。
2.2.2 總巰基
MP中巰基含量豐富,其暴露在氧化環(huán)境中易被轉(zhuǎn)變成分子內(nèi)或分子間二硫鍵及其他相關(guān)化合物,使MP中總巰基含量下降,進(jìn)而對(duì)蛋白的結(jié)構(gòu)和功能性質(zhì)造成影響[25]。如表1所示,未氧化MP中的總巰基含量為60.35 nmol/mg,與先前研究報(bào)道的值相似[14,19]。當(dāng)MP暴露于脂質(zhì)氧化體系中,隨著OLA濃度的增加,蛋白的總巰基含量穩(wěn)步下降,與未氧化MP相比,OLA濃度為10.0 mmol/L時(shí),總巰基含量下降約14.1%。類似的氧化引起MP總巰基含量降低的研究在各種氧化體系中已被廣泛報(bào)道[26-27]。
2.2.3 自由氨基
賴氨酸殘基的ε-NH2基團(tuán)在氧化過(guò)程中通過(guò)脫氨作用轉(zhuǎn)化為羰基,然后形成的羰基與氨基反應(yīng)生成席夫堿,進(jìn)而導(dǎo)致游離氨基的含量持續(xù)下降[28]。如表1所示,MP暴露于脂肪氧化體系后游離氨含量顯著下降(P<0.05)。與未氧化MP相比,經(jīng)過(guò)不同濃度OLA處理后(0.5、1.0、3.0和10.0 mmol/L)MP樣品自由氨基含量分別減少了15.2%、14.4%、17.6%和22.5%。脂質(zhì)氧化導(dǎo)致了MP中羰基的形成、巰基和游離氨基的損失,表明脂質(zhì)氧化產(chǎn)生的ROS對(duì)蛋白質(zhì)具有廣泛的氧化攻擊作用。
2.3 MP內(nèi)源性色氨酸熒光強(qiáng)度的變化
由于色氨酸殘基對(duì)周圍微環(huán)境的敏感性,內(nèi)源性色氨酸熒光特性通常用于反映蛋白質(zhì)結(jié)構(gòu)的變化[29]。當(dāng)?shù)鞍踪|(zhì)處于折疊狀態(tài)時(shí),MP具有較大的熒光強(qiáng)度和較短的最大發(fā)射波長(zhǎng);當(dāng)?shù)鞍踪|(zhì)結(jié)構(gòu)展開(kāi)時(shí),色氨酸殘基暴露于極性環(huán)境中,其熒光強(qiáng)度降低、最大發(fā)射波長(zhǎng)紅移(變長(zhǎng))。如圖1所示,隨著OLA濃度的增加,MP最大發(fā)射波長(zhǎng)處的熒光強(qiáng)度逐漸降低,特別是當(dāng)OLA濃度超過(guò)1.0 mmol/L時(shí)(P<0.05)。與此同時(shí),與未氧化MP相比,當(dāng)OLA濃度增加到10.0 mmol/L時(shí),MP的最大發(fā)射波長(zhǎng)從328.5 nm變?yōu)?29.6 nm。推測(cè)可能的原因是隨著氧化的進(jìn)行,蛋白質(zhì)結(jié)構(gòu)逐漸展開(kāi),MP中的色氨酸殘基等熒光基團(tuán)暴露在極性環(huán)境中,導(dǎo)致MP樣品的色氨酸熒光強(qiáng)度降低。
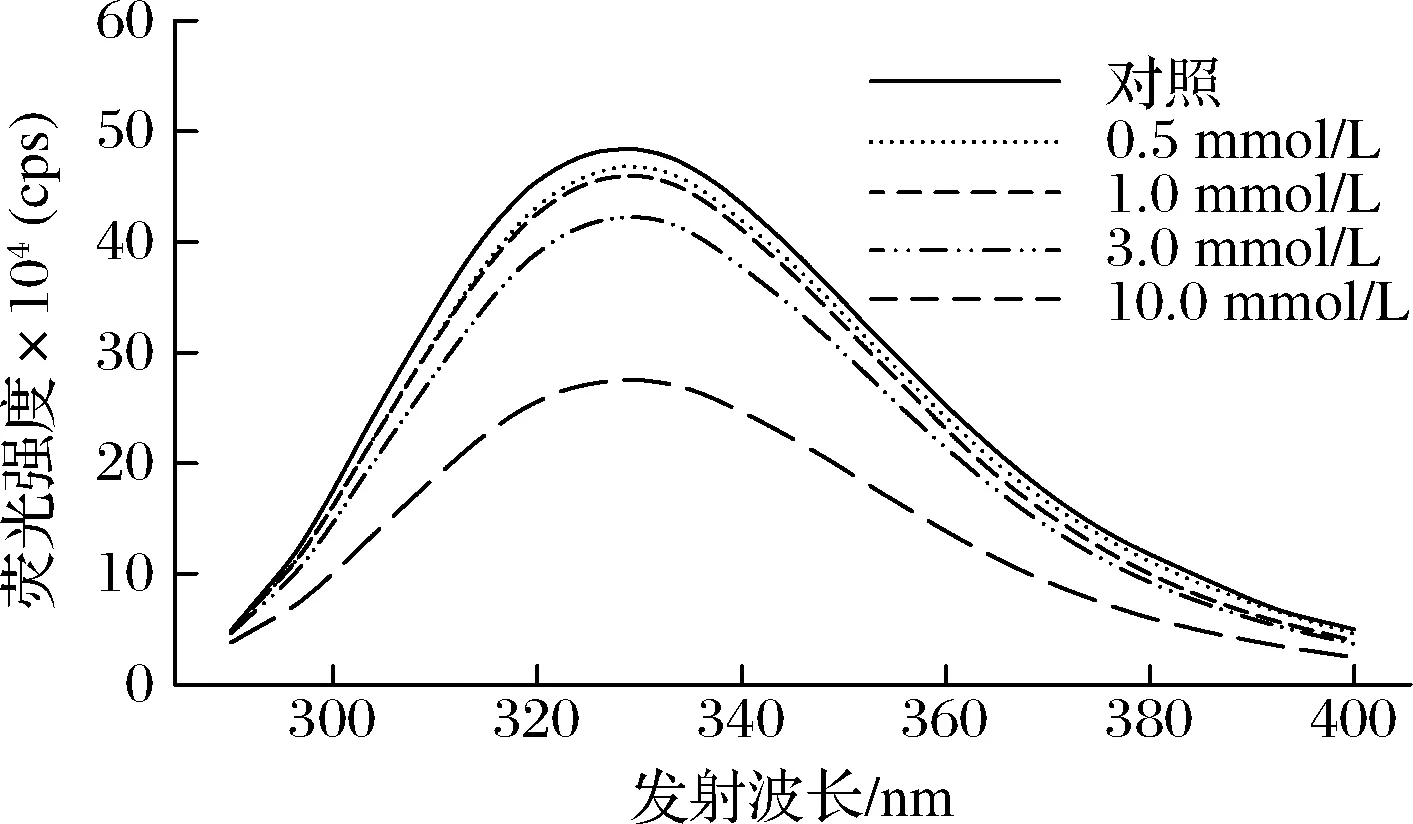
圖1 不同OLA濃度下MP的色氨酸熒光強(qiáng)度變化Fig.1 Changes in tryptophan fluorescence intensity of myofibrillar protein at different concentrations of OLA
2.4 MP樣品Zeta電位和粒徑的變化
Zeta電位反映了MP溶液中蛋白粒子間的靜電相互作用,它與MP溶液的穩(wěn)定性密切相關(guān)。本研究中MP樣品的Zeta電位均為負(fù)值(圖2-a),這與MP溶液的pH值高于蛋白等電點(diǎn)有關(guān),陰離子主要來(lái)自于蛋白質(zhì)顆粒表面的酸性氨基酸殘基(如天冬氨酸、谷氨酸等)。
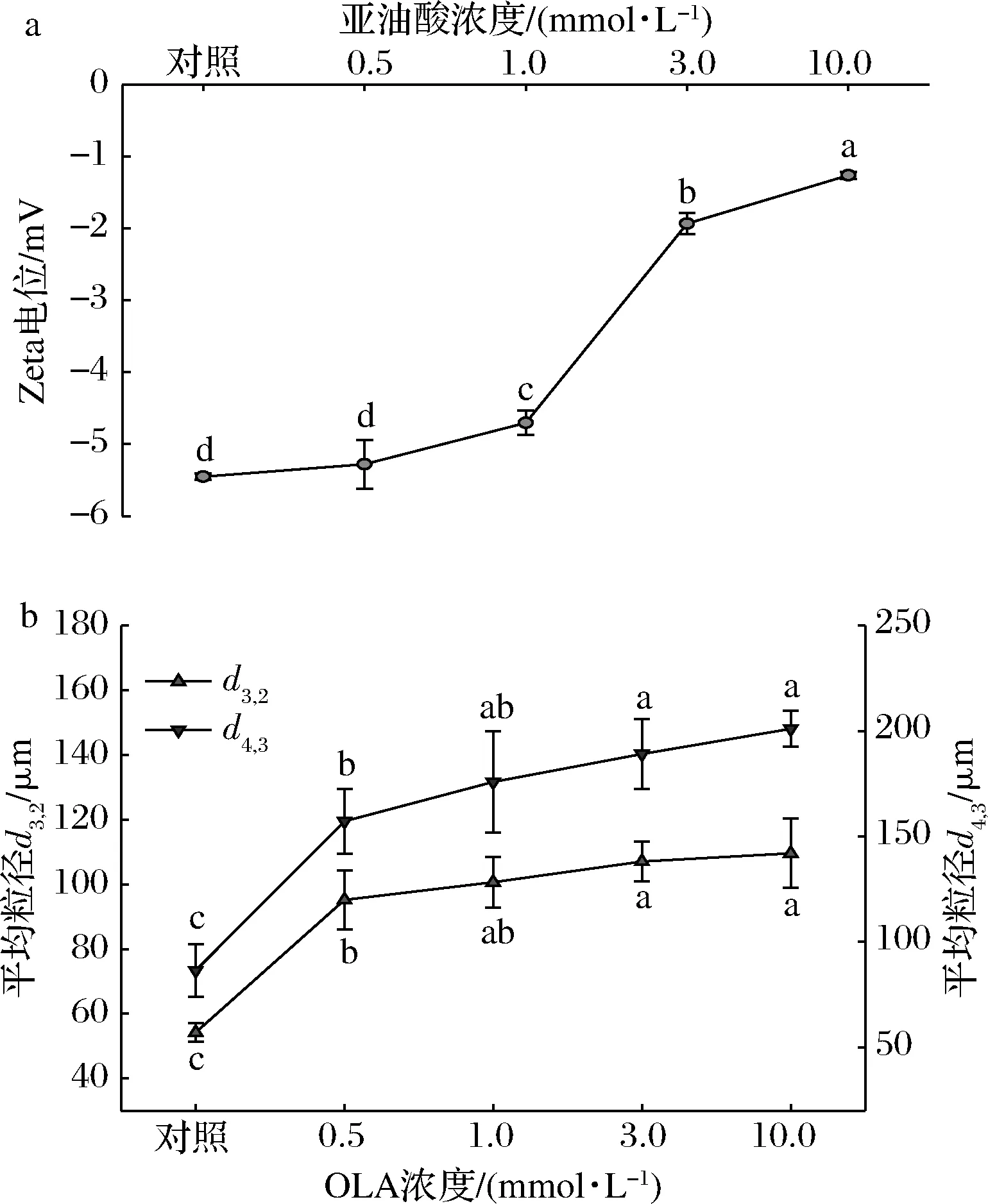
a-Zeta電位;b-粒徑圖2 不同OLA濃度下MP的Zeta電位和粒徑變化Fig.2 Changes in Zeta potential and mean particle diameter of myofibrillar protein at different concentrations of OLA
與未氧化MP相比,隨著OLA濃度的增加,MP的Zeta電位絕對(duì)值不斷減小,說(shuō)明蛋白粒子間的靜電作用力隨著氧化程度的增強(qiáng)而不斷減弱。
如圖2-b所示,通過(guò)測(cè)定MP樣品的平均粒徑d3,2和d4,3來(lái)評(píng)估氧化誘導(dǎo)的蛋白聚集行為。從圖中可以看到,隨著OLA濃度的增大,MP樣品的平均粒徑不斷增大。結(jié)合前文MP樣品溶液的內(nèi)源性色氨酸熒光強(qiáng)度以及Zeta電位結(jié)果發(fā)現(xiàn):氧化導(dǎo)致蛋白質(zhì)去折疊化,暴露出帶電荷的氨基酸殘基與氧化產(chǎn)物反應(yīng)生成電中性物質(zhì),從而使蛋白粒子間靜電相互作用減弱、蛋白質(zhì)分子間的交聯(lián)聚集加劇,引起蛋白平均粒徑的增加。這一結(jié)果與周非白[30]關(guān)于羥自由基氧化體系誘導(dǎo)的MP氧化以及BAO等[31]使用不同濃度次氯酸誘導(dǎo)蛋白氧化實(shí)驗(yàn)得到的MP平均粒徑變化趨勢(shì)相似。
2.5 MP溶解度的變化
MP的溶解度反映了蛋白質(zhì)交聯(lián)、聚集程度的變化。脂質(zhì)氧化程度對(duì)MP溶解度的影響如表1所示,未經(jīng)氧化處理的MP溶解度為43.35%,而當(dāng)MP暴露于脂質(zhì)氧化體系后蛋白溶解度持續(xù)降低(P<0.05)。與未氧化MP相比,在OLA濃度逐步增加至0.5、1.0、3.0、10.0 mmol/L時(shí),MP的溶解度分別下降了12.1%、7.2%、16.8%和43.3%。推測(cè)氧化后MP溶解度的降低主要與以下因素有關(guān):疏水基團(tuán)的暴露(圖1)、二硫鍵和其他共價(jià)鍵的形成,這些變化導(dǎo)致蛋白質(zhì)之間的相互作用增強(qiáng),分子間的交聯(lián)聚集加劇(圖2-b),從而降低了蛋白樣品的溶解度。
2.6 SDS-PAGE
不同濃度OLA處理下MP的SDS-PAGE圖譜如圖3所示。在非還原條件下(圖3-a,-DTT),隨著OLA濃度的增加,肌球蛋白重鏈(myosin heavy chain,MHC)和肌動(dòng)蛋白(actin)條帶強(qiáng)度穩(wěn)步下降,同時(shí)在濃縮膠的頂部觀察到高分子質(zhì)量聚合物的聚集,這一結(jié)果進(jìn)一步解釋了亞油酸誘導(dǎo)的脂質(zhì)氧化導(dǎo)致蛋白質(zhì)溶解度降低的原因。如圖3-b(+DTT)所示,在還原條件下堆積在濃縮膠頂端的聚合物幾乎完全消失,MHC和actin條帶強(qiáng)度明顯增強(qiáng),這一結(jié)果意味著氧化誘導(dǎo)形成的聚合物主要與MHC和肌動(dòng)蛋白有關(guān),同時(shí)也表明脂質(zhì)氧化引起的MP總巰基含量的下降主要是由于二硫鍵的形成[26,32]。
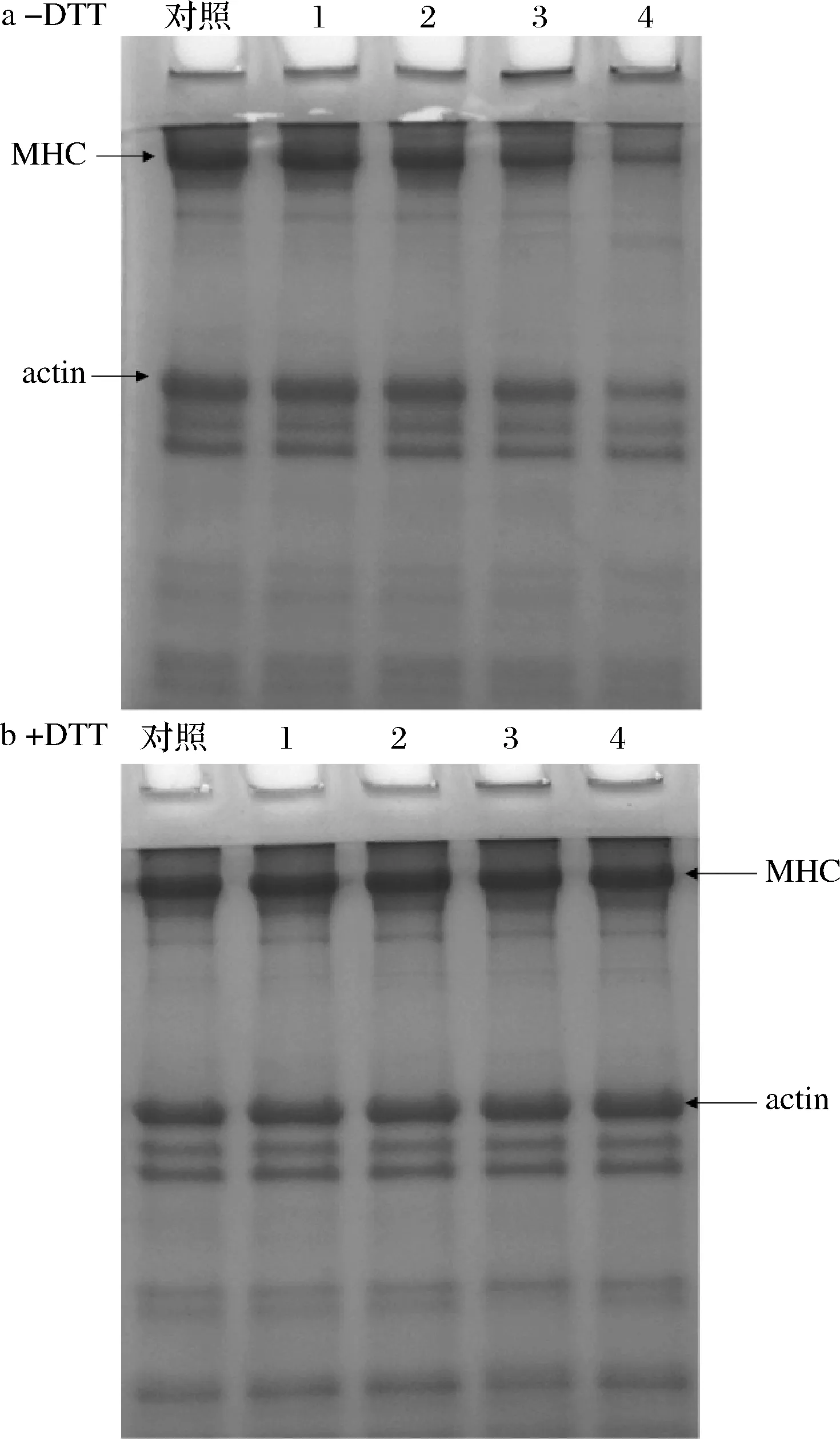
1-0.5 mmol/L;2-1.0 mmol/;3-3.0 mmol/L;4-10.0 mmol/L圖3 不同OLA濃度下MP SDS-PAGE圖譜的變化Fig.3 Changes of SDS-PAGE patterns of myofibrillar protein at different concentrations of OLA
2.7 MP凝膠性能的變化
2.7.1 動(dòng)態(tài)流變行為
MP懸浮液的流變特性可以反映蛋白質(zhì)氧化對(duì)凝膠形成過(guò)程以及凝膠彈性的影響。不同氧化處理的MP懸浮液在加熱過(guò)程中的流變規(guī)律如圖4所示。未氧化MP樣品呈典型的G′曲線,在49 ℃左右達(dá)到一個(gè)過(guò)渡峰,在53 ℃左右出現(xiàn)一個(gè)低谷,隨后在53~75 ℃穩(wěn)定上升。相關(guān)報(bào)道表明G′在49 ℃左右的第一個(gè)轉(zhuǎn)變峰是由肌球蛋白頭部的變性和聚集引起的[33],而約53 ℃出現(xiàn)的波谷則與輕酶解肌球蛋白變性有關(guān),此后G′的持續(xù)增加則歸因于永久性和不可逆肌球蛋白絲或復(fù)合物的形成[33-34]。OLA的存在顯著降低了初始的G′和過(guò)渡峰G′值的大小,然而最終G′值并沒(méi)有顯著變化。
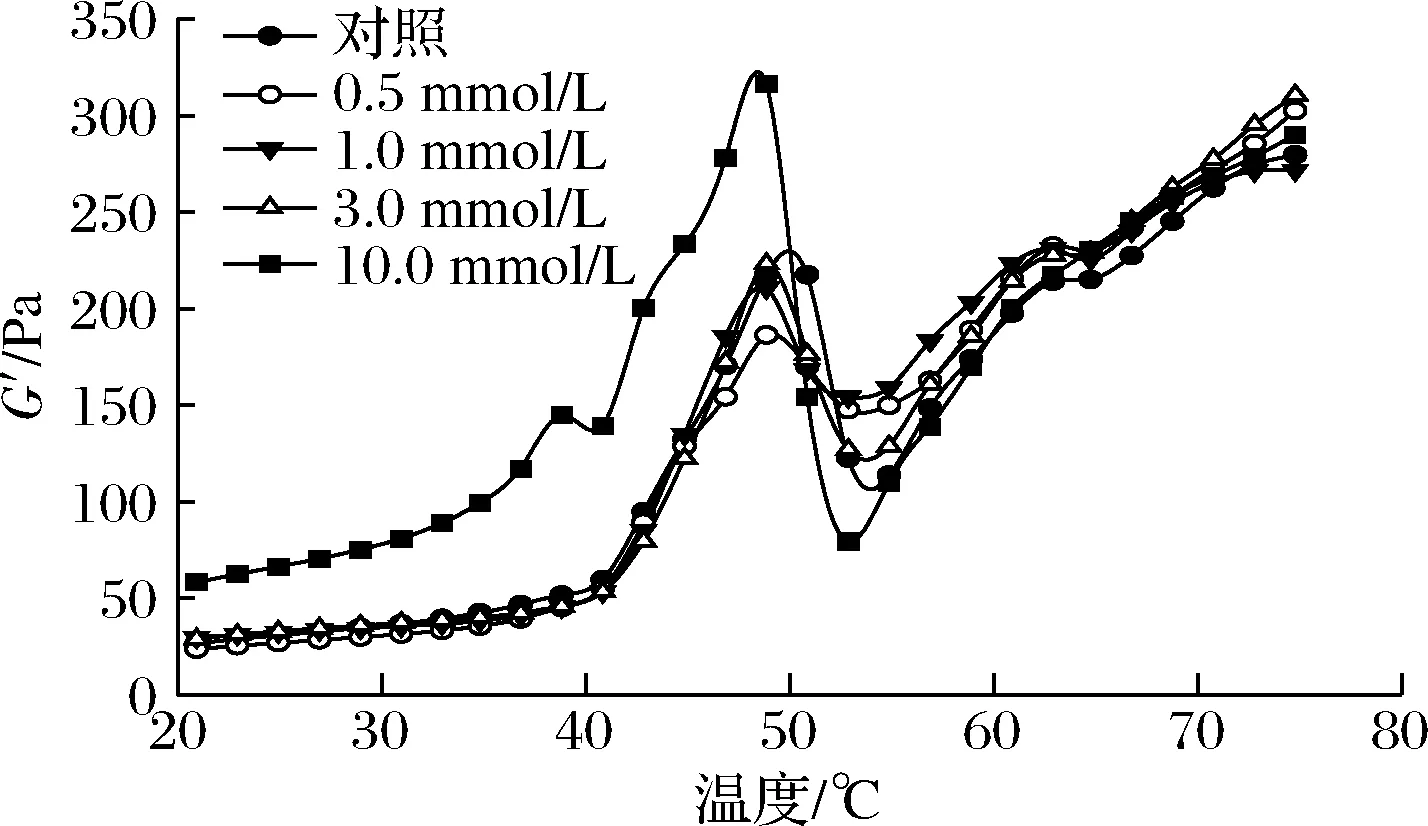
圖4 不同OLA濃度下MP的流變性能Fig.4 Rheological properties of myofibrillar protein at different concentrations of OLA
亞油酸誘導(dǎo)的脂質(zhì)氧化導(dǎo)致蛋白官能團(tuán)的修飾、蛋白質(zhì)構(gòu)象的變化以及MP聚集等現(xiàn)象的發(fā)生,從而改變了MP在加熱過(guò)程中的動(dòng)態(tài)流變行為。
2.7.2 蒸煮損失、凝膠強(qiáng)度和白度
如表2所示,隨著OLA濃度的增加,MP凝膠的蒸煮損失逐漸上升。與未氧化MP相比,在向MP樣品分別添加0.5、1.0、3.0和10.0 mmol/L的OLA后,MP凝膠的蒸煮損失分別增加46.6%、73.8%、78.6%和131.1%(P<0.05)。與蒸煮損失的變化趨勢(shì)相反,MP的凝膠強(qiáng)度隨著OLA濃度的增加而逐漸降低,經(jīng)0.5、1.0、3.0和10.0 mmol/L的OLA處理后,MP凝膠的初始斷裂力分別為對(duì)照組的82.1%、76.2%、75.0%和73.8%。推測(cè)隨著氧化強(qiáng)度的增加,MP分子間的交聯(lián)聚集加劇、溶解度不斷下降,這種情況影響了MP均勻牢固的凝膠網(wǎng)絡(luò)結(jié)構(gòu)的形成,造成了MP蒸煮損失的增加以及凝膠強(qiáng)度的降低[19,35]。UTRERA等[36]研究發(fā)現(xiàn)氧化導(dǎo)致蛋白質(zhì)羰基化,從而改變蛋白質(zhì)的電荷特性,導(dǎo)致肌肉蛋白質(zhì)的溶解度、保水性和凝膠特性降低,與本研究結(jié)果一致。
已有文獻(xiàn)報(bào)道表明蛋白的凝膠白度與蛋白質(zhì)的變性程度相關(guān)[37]。如表2所示,凝膠白度隨OLA濃度的增加而顯著降低,經(jīng)過(guò)0.5、1.0、3.0和10.0 mmol/L的OLA處理后的樣品凝膠白度分別為對(duì)照組的98.8%、98.4%、97.0%和95.5%。
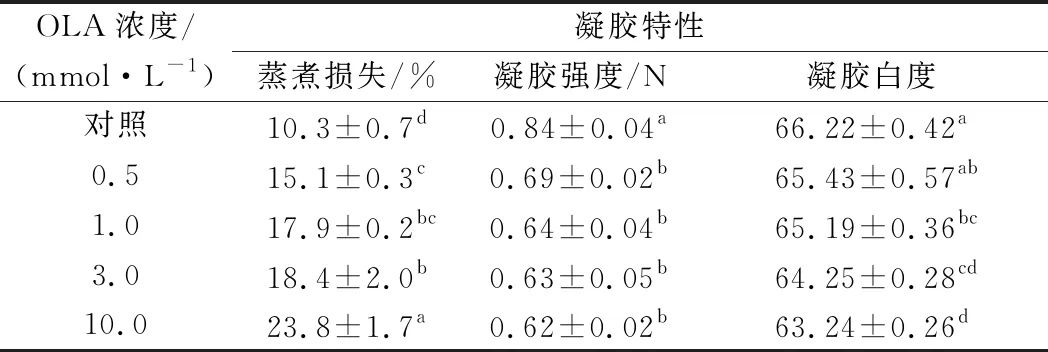
表2 不同OLA濃度下MP的凝膠性能變化Table 2 Changes in gelling properties of myofibrillar protein at different concentrations of OLA
周非白[30]研究發(fā)現(xiàn)隨著過(guò)氧自由基含量的增加,MP的凝膠白度顯著降低,與本研究結(jié)果一致。XIA等[38]發(fā)現(xiàn)豬肉MP的凝膠白度隨肌肉冷凍-解凍循環(huán)次數(shù)的增加而顯著降低,作者認(rèn)為這一現(xiàn)象與脂質(zhì)氧化有關(guān)。而在真實(shí)的肉類體系中,ESTéVEZ等[39]發(fā)現(xiàn)法蘭克福香腸的顏色與肌肉蛋白的氧化直接相關(guān)。因此,本研究中MP凝膠白度的降低應(yīng)與脂質(zhì)氧化和蛋白氧化相關(guān)。
2.7.3 MP凝膠的微觀結(jié)構(gòu)
蛋白質(zhì)凝膠微觀結(jié)構(gòu)的表征是研究MP凝膠特性的重要手段。不同OLA濃度對(duì)MP熱誘導(dǎo)凝膠微觀結(jié)構(gòu)的影響如圖5所示,未氧化MP形成的凝膠網(wǎng)絡(luò)結(jié)構(gòu)致密,微孔分布均勻。而OLA的添加對(duì)MP凝膠微觀結(jié)構(gòu)的影響主要與其濃度相關(guān),低濃度OLA的添加(0.5~1.0 mmol/L)對(duì)MP凝膠的微觀結(jié)構(gòu)沒(méi)有顯著影響,而當(dāng)OLA濃度從3.0 mmol/L增加到10.0 mmol/L時(shí),MP凝膠的微觀結(jié)構(gòu)發(fā)生顯著變化。當(dāng)OLA濃度達(dá)到10.0 mmol/L時(shí),MP凝膠的網(wǎng)絡(luò)結(jié)構(gòu)變得粗糙且不規(guī)則,孔隙變大,凝膠網(wǎng)絡(luò)結(jié)構(gòu)受到嚴(yán)重破壞。而MP凝膠松散、不規(guī)則的結(jié)構(gòu)也導(dǎo)致了MP凝膠持水性能和凝膠強(qiáng)度的降低。加熱前MP的高強(qiáng)度氧化引起的蛋白過(guò)度聚集和溶解度降低是導(dǎo)致MP凝膠性能下降以及微觀結(jié)構(gòu)粗糙不規(guī)則的原因。
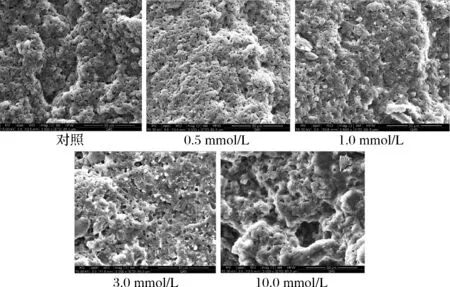
圖5 不同OLA濃度下MP凝膠的微觀結(jié)構(gòu)圖Fig.5 Microstructure of myofibrillar protein gels at different concentrations of OLA
2.7.4 拉曼光譜分析
760 cm-1(I760)附近的拉曼譜帶歸一化強(qiáng)度變化歸屬于色氨酸殘基的伸縮振動(dòng),其對(duì)色氨酸殘基周圍的微環(huán)境變化極其敏感,因此可用于研究MP凝膠的疏水相互作用[40]。如表3所示,與未氧化MP凝膠相比,經(jīng)過(guò)0.5 mmol/L的OLA處理后MP凝膠的I760值增加了約41.8%(P<0.05),這一結(jié)果表明輕度氧化增強(qiáng)了MP凝膠的疏水相互作用,然而隨著OLA濃度的進(jìn)一步增加,MP凝膠的I760值逐漸降低,表明過(guò)度氧化減弱了MP凝膠的疏水相互作用。
830和850 cm-1酪氨酸雙譜帶的相對(duì)強(qiáng)度(I850/I830)變化反應(yīng)了苯環(huán)上酚羥基(—OH)的狀態(tài)和氫鍵的性質(zhì)[40]。當(dāng)雙峰的相對(duì)強(qiáng)度(I850/I830)比值較高(0.9~2.5),表明酪氨酸殘基暴露在水溶液或其他極性微環(huán)境中或者同時(shí)作為氫鍵受體以及中弱等氫鍵供體進(jìn)行傳導(dǎo),而較低的相對(duì)比值則表明酪氨酸殘基埋藏在疏水環(huán)境中或僅作為氫鍵供體來(lái)增強(qiáng)氫鍵作用[21]。如表3所示,I850/I830的比值在0.982~1.036,這說(shuō)明酪氨酸殘基主要暴露在水溶液中并同時(shí)參與了中、弱氫鍵作用。需要注意的是,隨著OLA濃度的增加,I850/I830的比值由0.982增加到1.036,表明隨著氧化程度的加深,酪氨酸殘基的苯環(huán)上更多的—OH暴露于水環(huán)境中并與水分子形成氫鍵[41]。換言之,MP凝膠中蛋白-蛋白氫鍵隨著氧化程度的增加而減少。

表3 不同OLA濃度下MP凝膠I760的歸一化強(qiáng)度和I850/I830比率Table 3 Normalized intensity of the 760 cm-1band and ratio of 850/830 cm-1 doublet bands of myofibrillar protein gels at different concentrations of OLA
2.7.5 MP凝膠的體外消化測(cè)定
使用模擬胃和腸道消化的消化酶(胃蛋白酶、胰酶)來(lái)測(cè)定不同OLA濃度下MP熱誘導(dǎo)凝膠的消化率,結(jié)果如表4所示。在胃蛋白酶消化階段,與未氧化MP熱誘導(dǎo)凝膠相比,凝膠制備前的氧化處理對(duì)其最終消化率并沒(méi)有顯著性影響,當(dāng)OLA濃度過(guò)大時(shí)(3.0~10.0 mmol/L),MP凝膠的消化率有少許下降。在胰蛋白酶消化階段,與未氧化MP熱誘導(dǎo)凝膠相比,OLA濃度為0.5 mmol/L時(shí),蛋白在180 min時(shí)的消化率無(wú)顯著變化(P>0.05),而當(dāng)OLA濃度>0.5 mmol/L時(shí),MP凝膠的消化率顯著降低。這一結(jié)果也與周非白[30]在研究氧化對(duì)MP體外胃腸道消化率的結(jié)果類似,作者認(rèn)為過(guò)度氧化條件下蛋白形成聚集體,同時(shí)造成了氨基酸的損失,因此導(dǎo)致蛋白體外消化率的降低。而曹云剛[22]研究氧化及沒(méi)食子酸添加對(duì)MP凝膠體外消化率的影響時(shí)發(fā)現(xiàn),加熱前的氧化處理對(duì)MP凝膠的消化率無(wú)顯著影響,上述結(jié)果不一致,可能與實(shí)驗(yàn)所用肉的種類、試劑的量比以及實(shí)驗(yàn)條件的不同有關(guān)。
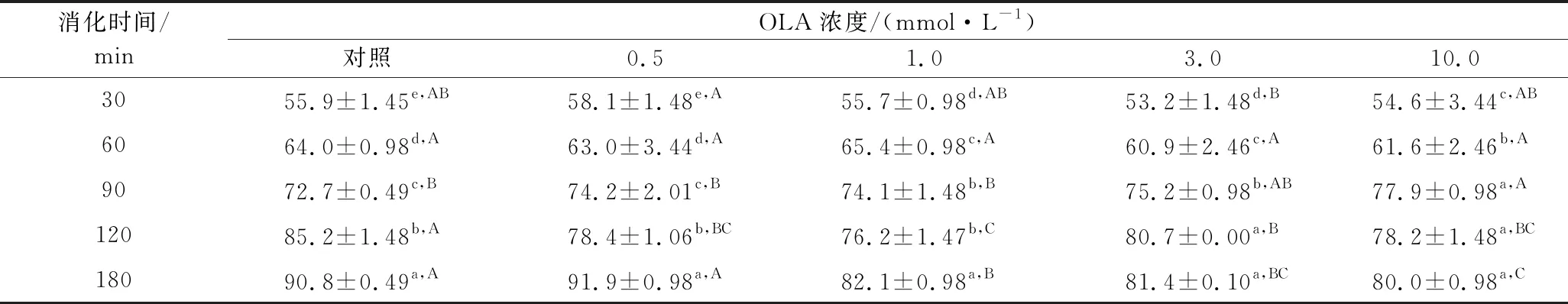
表4 不同OLA濃度下MP凝膠的體外消化率Table 4 In vitro digestibility of myofibrillar protein gels at different concentrations of OLA
3 結(jié)論
OLA誘導(dǎo)的蛋白氧化主要傾向于降低MP的凝膠性能,而這主要依賴于蛋白理化性質(zhì)以及結(jié)構(gòu)的改變。一般情況下,MP的氧化導(dǎo)致羰基的形成、總巰基和自由氨基含量的下降、Zeta電位絕對(duì)值的降低、蛋白質(zhì)的去折疊化以及由此導(dǎo)致的蛋白粒徑的增大和溶解度的下降。同時(shí),輕度的氧化對(duì)MP的膠凝特性沒(méi)有明顯影響,而加熱前MP的過(guò)度氧化會(huì)降低MP凝膠中蛋白顆粒間的疏水相互作用和氫鍵作用,從而降低了MP的凝膠性能以及蛋白凝膠的消化率。這些實(shí)驗(yàn)結(jié)果為研究OLA誘導(dǎo)的MP結(jié)構(gòu)變化及其與凝膠性能之間的關(guān)系提供了參考。
