電化學氮氣還原反應催化劑研究進展
2022-01-13 00:18:26路金偉白浩洋王雪飛
燕山大學學報 2022年1期
胡 婕,于 潔,路金偉,白浩洋,王雪飛
(1.燕山大學 環境與化學工程學院,河北 秦皇島 066004;2.燕山大學 河北省應用化學重點實驗室,河北 秦皇島 066004)
0 引言
隨著社會的蓬勃發展,化石燃料的消耗日益增多,探索可再生能源、尋找化石燃料的替代物受到了各界學者的廣泛關注。氨(NH3)是重要的無碳能源中間體之一,具有氫含量大、能量密度高、零碳排放等優點[1],并被廣泛用于化肥、樹脂、染料、藥物、炸藥、合成纖維和各種其他有機和無機化合物的制造[2-5],以滿足世界人口日益增長的需求。目前,NH3的生產有兩種方法,一種是固氮細菌通過固氮酶進行固氮的自然固氮,另一種是采用傳統的Haber-Bosch工藝進行固氮的工業固氮。然而,目前自然固氮已不能滿足世界工業快速增長的需求,同時,工業生產氨氣仍以H2和N2為原料進行高溫高壓(20~25 MPa,400~500 ℃)反應為主,需要大量的能源(約世界每年化石能源產出的1%~2%)并且產生大量的二氧化碳(約400 Mt)[6-8]。所以,設計和開發一種可持續的、綠色的固氮方法來替代傳統耗資耗能的Haber-Bosch工藝,有著十分重要的意義。
近幾十年來,人們一直致力于研究溫和條件下的固氮方法,如使用電化學或光化學系統來催化N2的還原。其中,電化學氮氣還原反應(nitrogen reduction reaction,NRR)在可再生電力驅動下以水為氫源產生NH3,是最理想的方法之一[9]。然而,這種方法仍然有許多問題沒有得到解決。首先,N2分子的吸附能力較差,且其中包含的惰性氮氮三鍵的裂解能較高(941 kJ/mol)[10],導致反應的動力學緩慢,因此NH3產率非常低。其次,NH3生產的選擇性較差,主要是因為在水溶液中,較低過電位下易發生競爭性析氫反應(hydrogen evolution reaction,HER)[11]導致法拉第效率較低。因此,為了降低惰性反應物的活化能障礙,加快電化學NRR過程,設計和合成各種具有NRR活性的電催化劑成為了諸多學者的研究熱點。
本文從電化學NRR的反應機理、NRR電催化劑以及相關催化劑的設計策略方面綜述了電化學NRR的研究進展,最后對本領域所面臨的挑戰以及今后的研究發展方向做了總結和展望,以期能給NRR電催化領域的學者一些啟發。
1 電化學氮氣還原反應機理
到目前為止,已經有很多團隊從實驗和密度泛函理論(density functional theory,DFT)計算的角度來研究水溶液中非均相催化劑的電化學NRR機制。有人提出,電化學NRR過程是一個復雜的質子耦合電子轉移(proton-coupled electron transfer,PCET)過程[12],所以它的反應機理也非常復雜。一般來說,電催化NRR過程有兩種基本機制,即解離機制(dissociative pathway)和締合機制(associative pathway),這兩種機制涉及的中間體不同(圖1)。對于解離機制,吸附的N2分子首先要經過氮氮三鍵的斷裂,這個過程需要吸收大量的能量,這就解釋了為什么遵循解離機制的Haber-Bosch過程需要非常苛刻的條件。
在締合機制中,在第一個NH3分子形成之前,被吸附的N2分子中兩個N原子保持相互結合的狀態,并在這種狀態下進行加氫過程[13]。根據N2分子吸附在催化劑表面的狀態以及加氫的順序,締合機制又可分為遠端加氫路徑、交替加氫路徑和酶機制。對于遠端加氫路徑和交替加氫路徑,N2分子垂直吸附在催化劑活性位點上,一個N原子與活性位點結合;酶機制則是N2分子水平吸附在催化劑活性位點上,兩個N原子都與活性位點結合。遠端加氫路徑中,遠離吸附端的遠端N原子優先氫化直到遠端NH3分子釋放,剩下的另一個N原子重復同樣的氫化過程產生第二個NH3分子。而交替加氫路徑則是兩個N原子伴隨PCET過程輪流交替加氫,兩個NH3分子在該路徑的最后一步連續釋放[14-15]。一般情況下,電化學NRR的限速步驟是加第一個H原子的過程,即由*N2轉化為*N2H(*表示N2吸附的活性位點)的過程,而有的催化劑能夠改變限速步驟[16],從而減小限速步驟的能壘,相應地,催化劑的性能就好。
最近,Abghoui和Skúlason提出了Mars-van Krevelen (MvK)機制,這種機制只適用于過渡金屬氮化物(transition metal nitrides,TMNs),比常規的解離機制和締合機制更有利于電化學NRR。在MvK機制中,TMNs表面的一個晶格氮原子還原為NH3,隨后電解液中的N2分子填充進產生的氮空位中,接著進行加氫過程。DFT計算結果證明,在潔凈的過渡金屬氮化物表面通過解離機制生成氨氣的活化障礙較大,且通過MvK機制預測的過電勢比通過締合機制預測的小[17]。Ren等[18]合成的氮化鉬(Mo2N)納米棒就是通過MvK機制進行電化學NRR的。在氮化鉬納米棒上進行的電化學NRR反應中,電位決定步驟(potential-determining step,PDS)所需的最大自由能(0.66 eV)比在二氧化鉬上所需的最大自由能(1.26 eV)小,因此,在相同的條件下氮化鉬作為NRR的電催化劑比二氧化鉬活性更強。
綜上所述,從電化學NRR機理推測,影響NRR電催化劑性能的關鍵因素為:1)催化劑表面N2分子的吸附與活化;2)電化學NRR的選擇性和HER;3)電化學NRR過程中催化劑表面的加氫過程。因此合成能夠促進N2分子的吸附與活化、抑制析氫反應并能夠降低電化學NRR過程中反應能壘的催化劑是使電催化NRR能夠替代Haber-Bosch工藝的關鍵。
2 電化學氮氣還原反應催化劑
常溫常壓條件下NRR的各種電催化劑,根據其使用成本和稀缺程度一般分為兩種:貴金屬基催化劑和非貴金屬基催化劑。本節討論了這兩種電催化劑在常溫常壓條件下的電催化NRR性能。表1和表2給出了不同NRR電催化劑的性能總結[19-25]。

表1 貴金屬催化劑的電化學NRR性能Tab.1 Performance of noble metal catalyst for electrochemistry NRR

表2 非貴金屬催化劑的電化學NRR性能Tab.2 Performance of non-noble metal catalyst for electrochemistry NRR
2.1 貴金屬催化劑


圖2 SA-Ag/NC上*N2和*NNH的端吸附構型Fig.2 End-on configurations of *N2 and *NNH on SA-Ag/NC
2.2 非貴金屬催化劑
貴金屬儲量低、價格高,嚴重限制了其應用。相比之下,過渡金屬儲量豐富,價格低廉。而且,由于過渡金屬具有特定的電子結構:具有未占據的d軌道、具有適當能量和對稱性的已占據軌道,因此可以作為合成NH3的電催化劑。
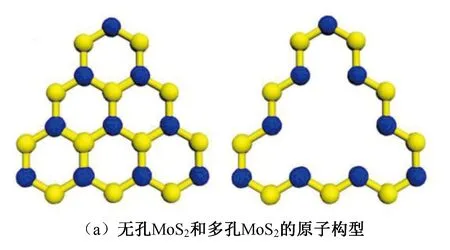
圖3 MoS2的原子構型以及N2吸附中間體的結構Fig.3 The atomic configuration of MoS2 and the structures of adsorbed intermediates of N2
除了鉬基催化劑,學者們還對Fe基催化劑[38-40]、Cu基催化劑[41]、Bi基催化劑[42]、MXene材料[43-44]等進行了研究,并取得了一定的成果。
過渡金屬基催化劑在實際應用中存在一些固有的缺陷:1)大多數過渡金屬催化劑由于與N2的結合較弱,對N2的激活能力不夠強;2)過渡金屬對析氫反應(HER)也有利;3)催化過程中可能會釋放過渡金屬,造成一些環境問題[45]。近年來,無金屬電催化劑由于其對質子的吸附能力弱,產氫性能低,在電化學NRR中引起了廣泛關注。目前研究較多的非金屬催化劑有碳基催化劑、磷基催化劑等。
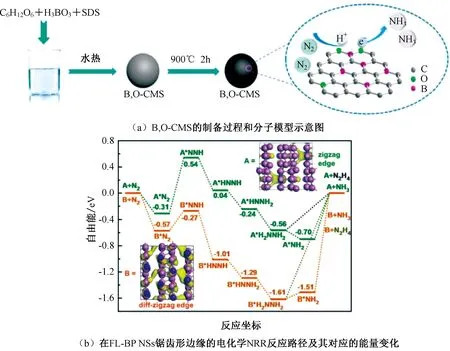
圖4 B,O-CMS的制備過程和分子模型示意圖以及在FL-BP NSs鋸齒形邊緣的NRR反應路徑及其對應的能量變化Fig.4 Schematic illustration of the fabrication process and molecular model of B,O-CMS,reaction pathways and the corresponding energy changes for the NRR on the zigzag-edge of FL-BP NSs
3 催化劑的設計策略
目前,常溫常壓條件下電催化NRR存在過電位高、法拉第效率低、NH3產率低等問題,嚴重阻礙了其發展。克服這些障礙的有效方法是設計高活性、耐用的NRR電催化劑。目前,常用的催化劑設計策略包括表/界面工程(構建異質結、形貌尺寸調控)、晶面調控與非晶化、缺陷工程(空位調控、雜原子摻雜)、構建仿生位點等[51-56]。
3.1 表/界面工程
電化學NRR還原反應通常發生在催化劑表面,所以催化劑的表面性質對其催化活性有較大的影響。通過表/界面工程調控催化劑的外觀形貌和電子結構來改善其表面性質是提高催化劑活性的有效策略。
3.1.1 形貌尺寸調控
3.1.2 構建異質結

圖5 CoPi/NPCS和CoPi/NPC的掃描電鏡圖以及MoS2/C3N4的高分辨透射電鏡圖Fig.5 SEM images of CoPi/NPCS and CoPi/NPC,HRTEM image of MoS2/C3N4
3.2 晶面調控與非晶化
合理調控晶面的原子排列和配位,最大限度地暴露活性位點是一種提高電催化劑活性的有效策略。研究表明,階梯式晶面是催化劑上良好的活性位點,電化學NRR過程中的中間體與階梯上的活性位點的結合比與平面上的更強。Bao等[61]發現由{730}晶面族(由(210)和(310)晶面組成)圍成的二十四面體金納米棒具有一定的電化學NRR性能,該催化劑暴露的階梯{730}晶面族提供了豐富的活性位點(圖6(a)),促進了N2的吸附和解離。在-0.2 V vs.RHE電位下得到了1.648 μg·h-1·cm-2的氨產率。
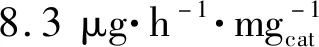
3.3 缺陷工程
3.3.1空位工程
營造空位使催化劑結構含有缺陷,是改善催化劑性能的有效策略之一。研究表明,各種空位的存在可以有效地調節電子結構、電荷輸運和表面吸附容量等催化劑的固有性質。因此,這些空位作為多相催化的主要吸附和活性位點,降低了活化能壘,促進了催化反應的進行。目前報道較多的空位有氮空位(nitrogen vacancies,NVs)、硫空位(sulphur vacancies,SVs)、氧空位(oxygen vacancies,OVs)等[65]。


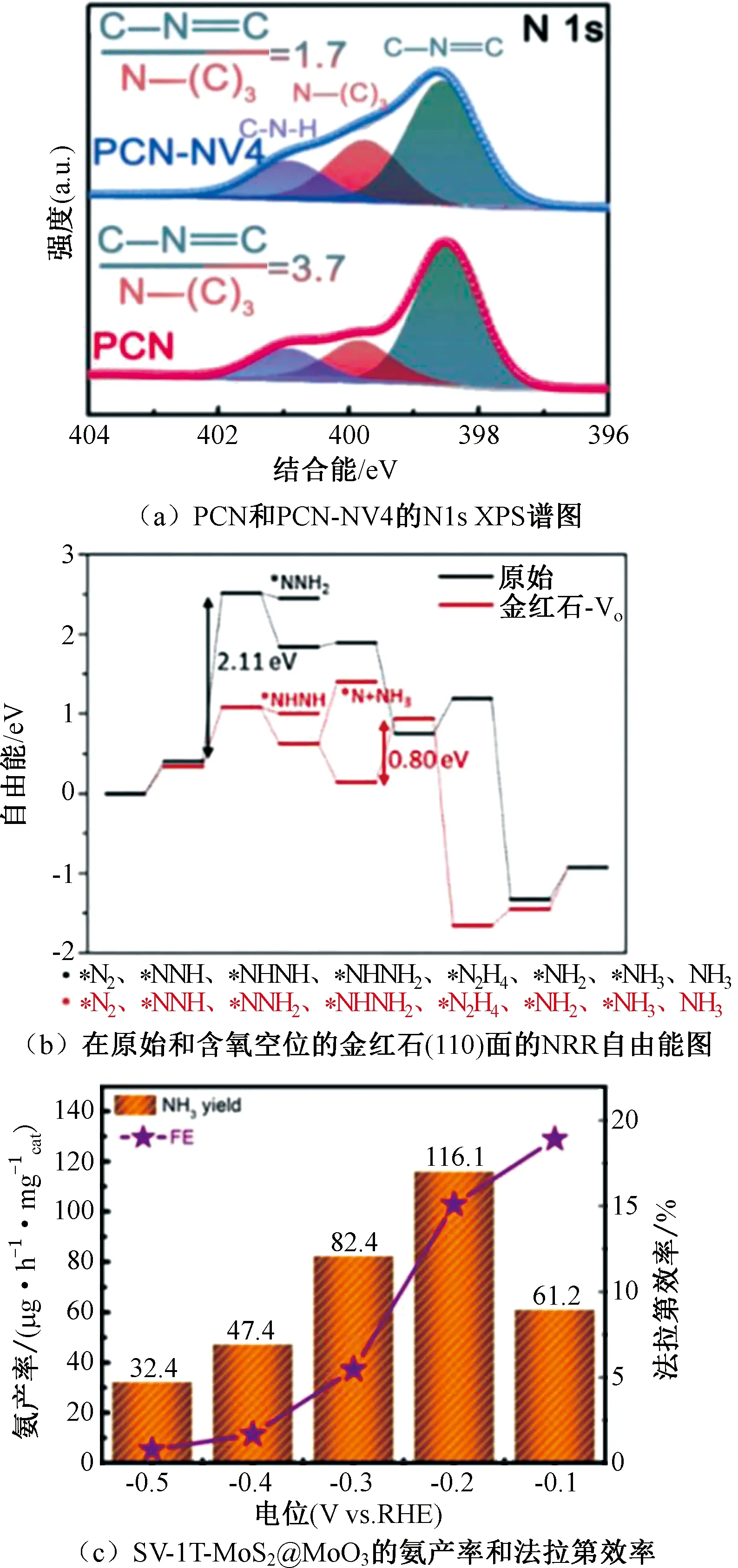
圖7 XPS譜圖、自由能圖以及NH3產率和法拉第效率圖Fig.7 XPS spectra、free energy diagram and the picture of NH3 yields and Faradaic efficiencies
3.3.2 雜原子摻雜
雜原子摻雜被認為是調整催化劑電子結構、使催化劑富含缺陷的另一種方法。該方法可以調節催化劑的電荷分布從而調節反應物、中間體和產物在催化劑表面的吸脫附過程。摻雜劑一般可分為兩類:非金屬摻雜劑和金屬摻雜劑。
研究表明,非金屬元素硼是一種重要的摻雜元素,Yu等[70]通過將硼酸和氧化石墨烯一起退火合成了硼摻雜石墨烯,摻雜硼原子的石墨烯保留了原來的sp2雜化和共軛平面結構,且硼的摻雜誘導石墨烯形成了電子缺陷,從而大大提高了其電催化NRR活性。DFT計算結果顯示,硼元素與石墨烯形成的BC3鍵既有利于N2的固定,也有利于NH3的脫附,是主要的活性位點(圖8(a))。該催化劑表現出較好的催化活性,其NH3產率和法拉第效率分別為9.8 μg·h-1·cm-2和10.8%。除此以外,Liu等[71]制備的硼摻雜金剛石、Lan等[72]制備的納米多孔硼、Yu等[73]制備的B、S共摻雜銠膜都具有較好的電化學NRR活性。除硼元素外,碳元素、氮元素、磷元素以及硫元素也經常作為摻雜劑對NRR電催化劑進行改性[74-78]。
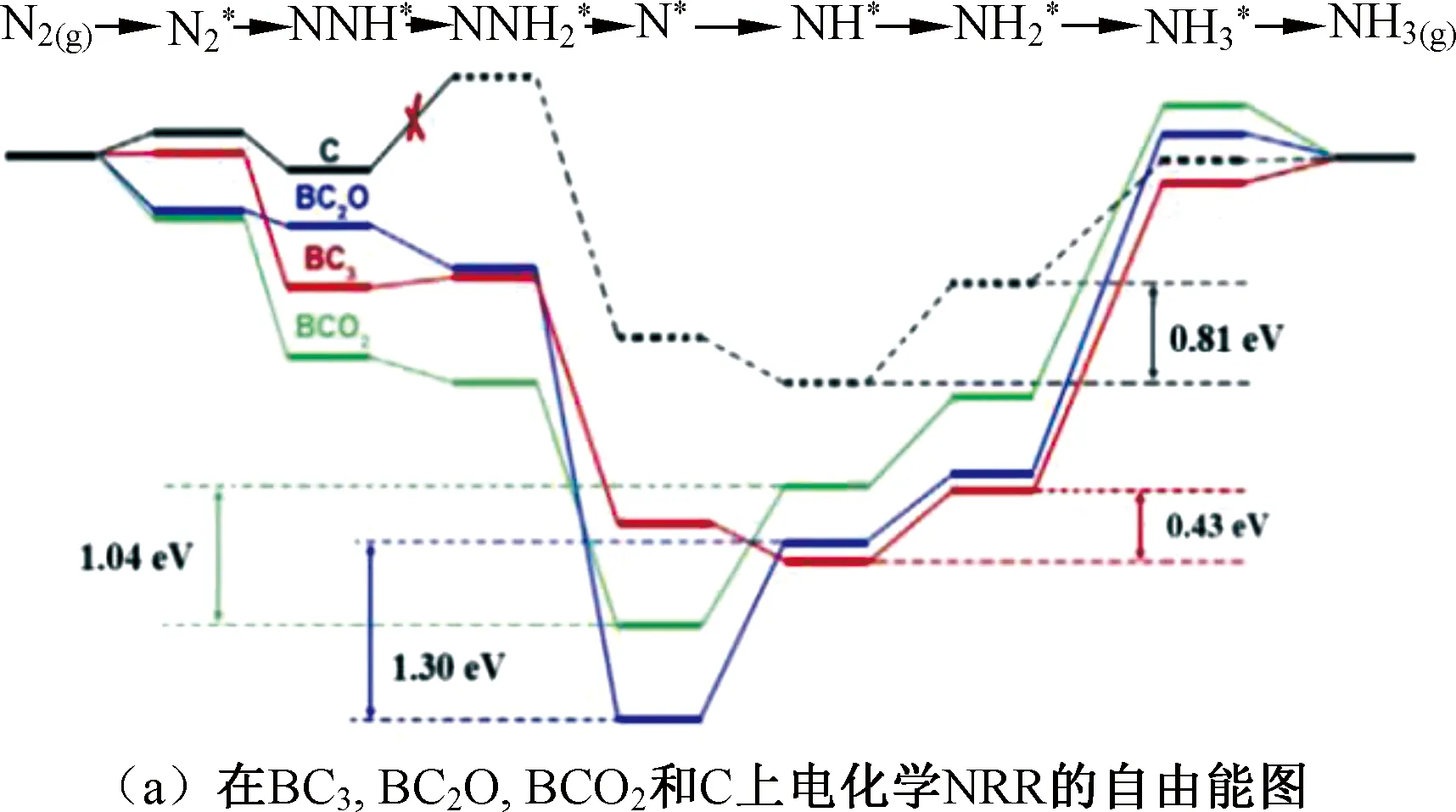
圖8 電化學NRR的自由能圖以及機理圖Fig.8 Free energy and mechanism diagrams of electrochemistry NRR
3.4 構建仿生位點
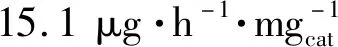


圖9 仿生電極-電解質設計的原理圖Fig.9 Schematic representation of the biomimetic electrode-electrolyte design
4 總結與展望
電化學NRR作為傳統Haber-Bosch工藝的替代無疑具有巨大的潛力,它具有反應條件溫和、成本低、零碳排放等優點,能有效緩解能源危機以及二氧化碳大量碳排放導致的全球變暖等問題。然而該方法依然存在一些問題需要解決,如催化劑活性低導致的NH3產率和法拉第效率低、催化劑的穩定性差等。因此,可以從以下幾個方面展開工作:
1)提升已有催化劑的催化活性。結合表/界面工程、晶面調控與非晶化、缺陷工程等方法增加催化劑表面的活性位點,改善N2的吸附與活化從而改善催化劑的催化活性。
2)開發新型電化學NRR催化劑。通過仿生、計算等策略有目標地進行催化劑的設計與合成,開發適合電化學NRR的新型催化劑。
3)提高NRR電催化劑的穩定性。一方面可以通過催化劑的設計與調控改善其物理化學性能,從而提高催化劑的穩定性;另一方面,明確催化劑的失活機制,確保活性位點的有效暴露也可以提高催化劑的穩定性。
猜你喜歡
四川勞動保障(2021年9期)2022-01-18 05:11:08
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
文苑(2018年21期)2018-11-09 01:23:06
中國衛生(2016年9期)2016-11-12 13:28:08
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國衛生(2015年9期)2015-11-10 03:11:12
中國衛生(2014年3期)2014-11-12 13:18:12
