α-芳基-α,β-不飽和羰基化合物的合成
2022-04-13 05:10:54董濤濤陳書升
浙江師范大學學報(自然科學版) 2022年2期
彭 勃, 董濤濤, 陳書升
(浙江師范大學 化學與生命科學學院,浙江 金華 321004)
α-芳基-α,β-不飽和羰基化合物作為一類重要的有機官能片段,廣泛存在于藥物及天然產物中[1-8].香料類化妝品中的可卡醛[2]、抗癌藥物黃豆黃素[3]、吡侖帕奈[4]、葛根素[7]及天然產物雞豆黃素[8]中都含有α-芳基-α,β-不飽和羰基片段(見圖 1).此外,α-芳基-α,β-不飽和羰基化合物是有價值的有機合成中間體,例如α-芳基-α,β-不飽和羰基化合物通過官能團衍生化,可以實現多取代吲哚的合成[9-11]、全合成的應用[12-14]及α-芳基-α,β-不飽和羰基化合物進一步偶聯、加成等反應實現α,β-不飽和羰基化合物雙官能團化[15].因此,α-芳基-α,β-不飽和羰基的合成是有機合成化學的重要研究目標之一.鑒于此,本文將介紹構建α-芳基-α,β-不飽和羰基化合物的兩類合成方法.
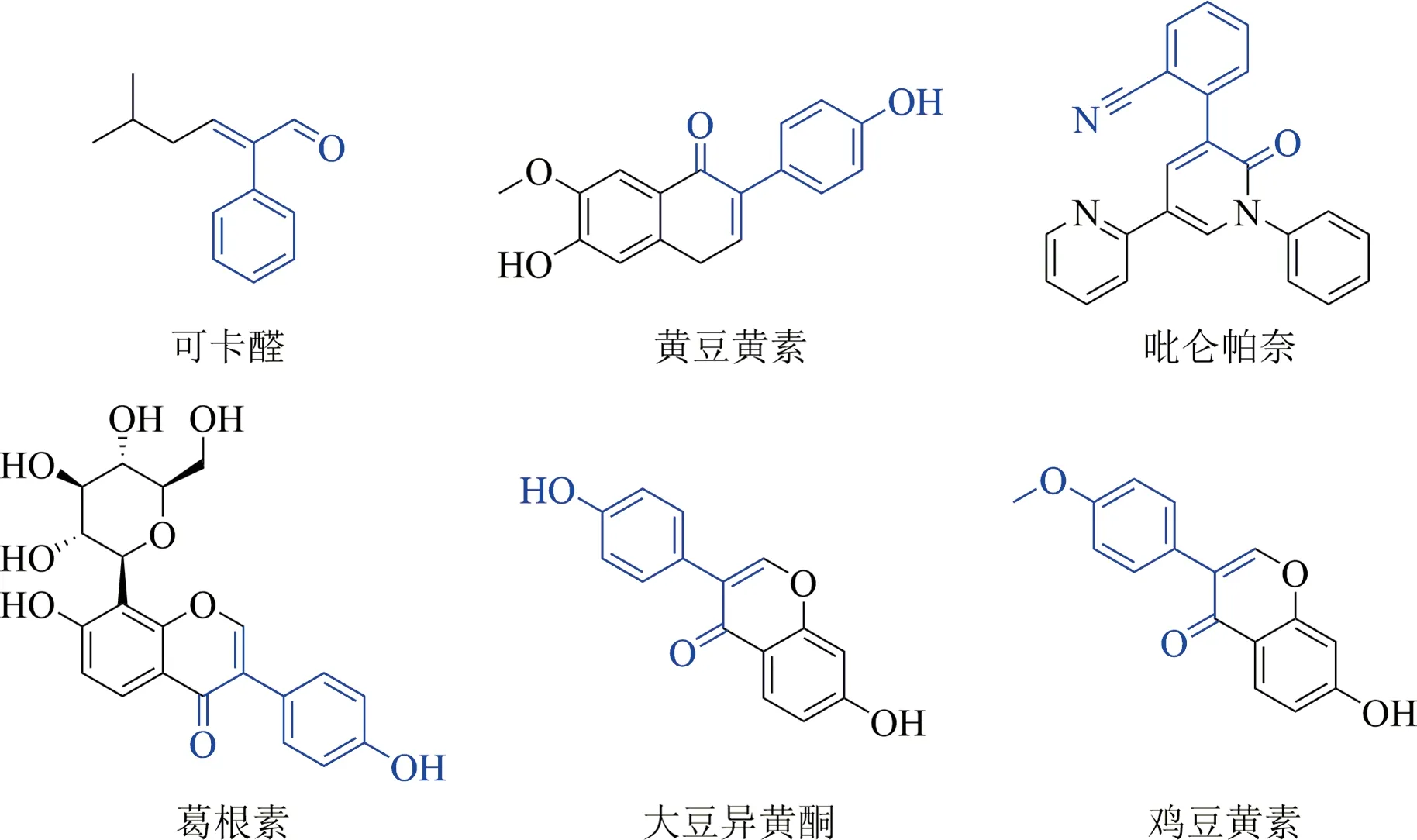
圖1 含α-芳基-α,β-不飽和羰基片段的生物活性分子
1 間接合成法構建α-芳基-α,β-不飽和羰基化合物
1.1 α-鹵代-α,β-不飽和羰基化合物與芳基金屬試劑偶聯
反應通式如圖2式(1)所示,α,β-不飽和羰基化合物與鹵素分子作用合成α-鹵代-α,β-不飽和羰基化合物,隨后在鈀催化劑的催化下與芳基金屬試劑偶聯得到α-芳基-α,β-不飽和羰基化合物;或者由α-金屬-α,β-不飽和羰基化合物和鹵素分子反應合成α-鹵代-α,β-不飽和羰基化合物,并在鈀催化劑的作用下與芳基金屬試劑偶聯得到α-芳基-α,β-不飽和羰基化合物.
1992年,Johnson等[16]首先發展了2-環己烯酮在四氯化碳和吡啶溶劑中與碘單質反應生成2-碘-2-環己烯-1-酮,隨后2-碘-2-環己烯-1-酮與對甲苯基三丁基錫烷的催化偶聯反應,實現2-環己烯酮的α-芳基化,反應有8個化合物(產率為55%~95%),其中3a偶聯產率達到86%(見圖2式(2)).該類反應在后續10年里得到很大發展[17-20].
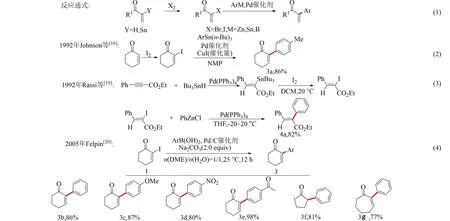
圖2 α-鹵代-α,β-不飽和羰基化合物與芳基金屬試劑偶聯的模式及部分實例
同年,Rassi等[19]報道了苯丙炔酸甲酯與三丁基錫烷在鈀催化的條件下選擇性地合成(E)-2-(三丁
基錫烷基)-3-苯基丙烯酸甲酯,隨后與碘單質反應得到(E)-2-碘-3-苯基丙烯酸甲酯,并最終與芳基鋅試劑偶聯得到(E)-2(對甲苯基)-3-苯基丙烯酸甲酯4a,偶聯產率82%(見圖2式(3)).雖然該反應能選擇性得到(E)-α-芳基-α,β-不飽和羰基化合物4a,但該反應的反應性較差,多步反應后總產率僅47%,反應底物范圍窄,僅給出4個化合物(收率56%~82%).隨后在2005年,Felpin[20]通過2-環己烯-1-酮的α碘代,制備2-碘-2-環己烯-1-酮,并與苯硼酸偶聯,實現了2-環己烯酮的α-芳基化(見圖2式(4)),偶聯產率86%.與Johnson反應[16]相比,Felpin反應突破了只能使用對甲基類底物的限制,并將底物范圍拓展到吸電子類底物,如3d(產率80%)和3e(產率98%).
1.2 α-金屬-α,β-不飽和羰基化合物與鹵代芳烴偶聯
反應通式如圖3式(5)所示,α,β-不飽和羰基化合物與金屬試劑反應生成α-金屬-α,β-不飽和羰基化合物,隨后在催化劑作用下與鹵代芳烴劑偶聯得到α-芳基-α,β-不飽和羰基化合物;或者通過α-鹵代-α,β-不飽和羰基化合物和鋅等金屬試劑反應合成相應的鋅試劑,并與鹵代芳烴偶聯得到α-芳基-α,β-不飽和羰基化合物.
1984年,Chang等[21]首次報道了α-汞-α,β-不飽和羰基化合物與碘代芳烴的偶聯反應,隨后該類反應得到了進一步的發展[22-25].1993年,Levin[23]使用丙炔酸甲酯與三丁基錫烷反應合成2-(三丁基錫烷基)-2-丙烯酸甲酯,并在四三苯基膦鈀與碘化亞銅催化作用下實現2-(三丁基錫烷基)-2-丙烯酸甲酯與對硝基碘苯的偶聯,制備2-(對硝基苯基)-丙烯酸甲酯5a,偶聯產率76%.該反應方法給出了10個化合物(產率42%~92%)(見圖3式(6)).1997年,Prasad等[24]報道了2-碘-環己烯基酮與金屬鋅反應生成鋅試劑,并在N,N-二甲基甲酰胺溶液中與對硝基碘苯偶聯得到3h,偶聯產率88%(見圖3式(8));2002年,Jalil等[25]報道了使用2-溴丙烯酸乙酯與金屬鋅反應制備的鋅試劑與碘苯的催化偶聯反應.與Levin反應[24]相比,Jalil偶聯反應產率高達98%,不僅如此,該反應突破此前對于含給電子基團底物產率低的局限,對該類底物也能達到98%的產率(見圖3式(7)).
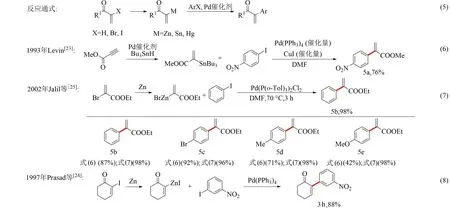
圖3 α-金屬-α,β-不飽和羰基化合物與鹵代芳烴偶聯反應與部分實例
1.3 α-鹵代-α,β-不飽和羰基化合物與鹵代芳烴偶聯
反應通式如圖4式(9)所示,α,β-不飽和羰基化合物與鹵素分子反應生成α-鹵代-α,β-不飽和羰基化合物,隨后在催化劑的作用下與鹵代芳烴劑偶聯得到α-芳基-α,β-不飽和羰基化合物.
2003年,Banwell等[9]首次報道了從2-環己烯酮出發,制備2-碘-2-環己烯-1-酮,隨后與對硝基碘苯發生偶聯反應生成2-(對硝基苯基)-2-環己烯-1-酮,實現2-環己烯酮α芳基化3i,偶聯反應產率85%(見圖4式(10)).
在2006年,Molander等[26]報道了使用肉桂醛制備2-溴-3-苯基-2-丙烯醛,并在碳酸鉀與鈀催化劑的作用下實現與芳基三氟硼酸鉀交叉偶聯反應得到6a,偶聯反應產率94%.但Molander等對該反應的底物范圍探索比較少,僅有4例化合物(產率87%~97%)(見圖4式(11)).
在2013年,Tasch等[27]使用鈀催化的鹵代芳烴與烯基溴化物的交叉偶聯反應構建α-芳基-α,β-不飽和羰基化合物6b.與Molander相似,Tasch等對于該反應的底物范圍較少,僅給出了3個化合物(產率65%~80%)(見圖4式(12)).
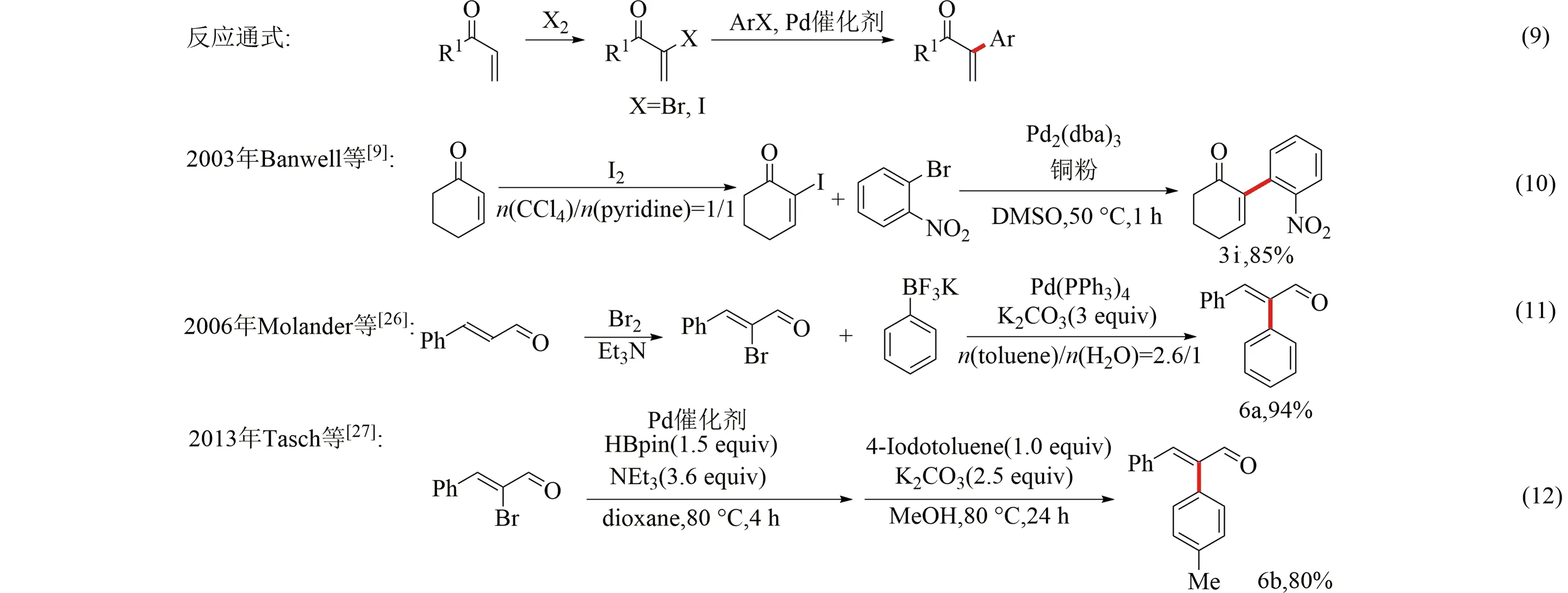
圖4 α-鹵代-α,β-不飽和羰基化合物與鹵代芳烴偶聯反應模式及部分實例
2 直接合成法構建α-芳基-α,β-不飽和羰基化合物
2.1 α,β-不飽和羰基化合物與芳基鉍試劑直接偶聯
2004年,Koech等[28]首次報道了在二氯甲烷與叔丁醇及二異丙基乙基胺反應條件下,以三丁基膦為催化劑,芳基鉍試劑8為芳基化試劑實現了2-環己烯酮(或2-環戊烯酮)7的α-芳基化反應.該反應條件溫和.反應對含給電子基團的底物的兼容性差,反應物9b產率僅44%(見圖5).
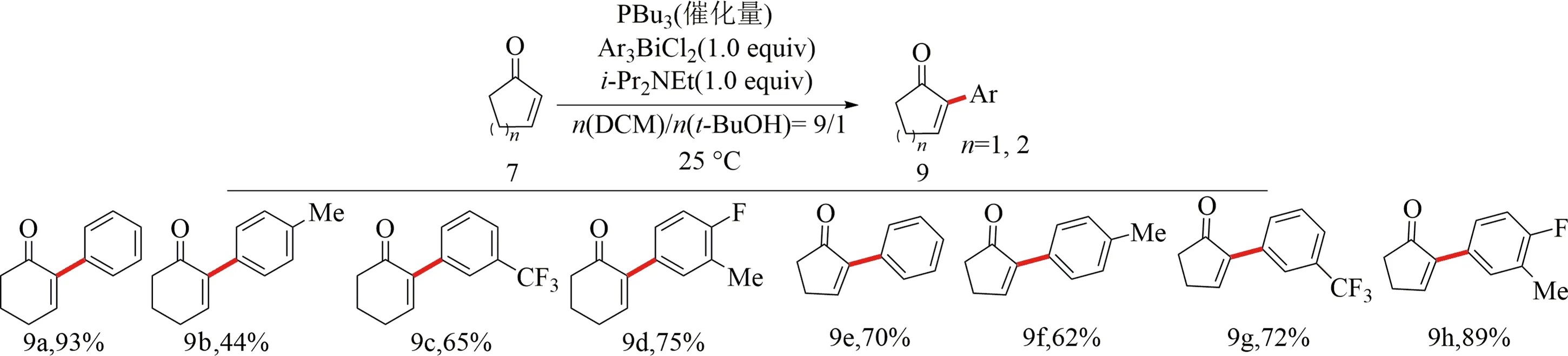
圖5 α,β-不飽和羰基化合物與芳基鉍試劑直接偶聯[28]
Koech提出以下反應機理:首先三丁基膦與2-環己烯酮發生Michael加成;得到烯醇中間體,隨后芳基鉍試劑與烯醇中間體的氧負離子結合;隨后脫除三丁基膦、氯化氫及二芳基氯化鉍;最終得到α芳基化的2-環己烯酮(見圖6).
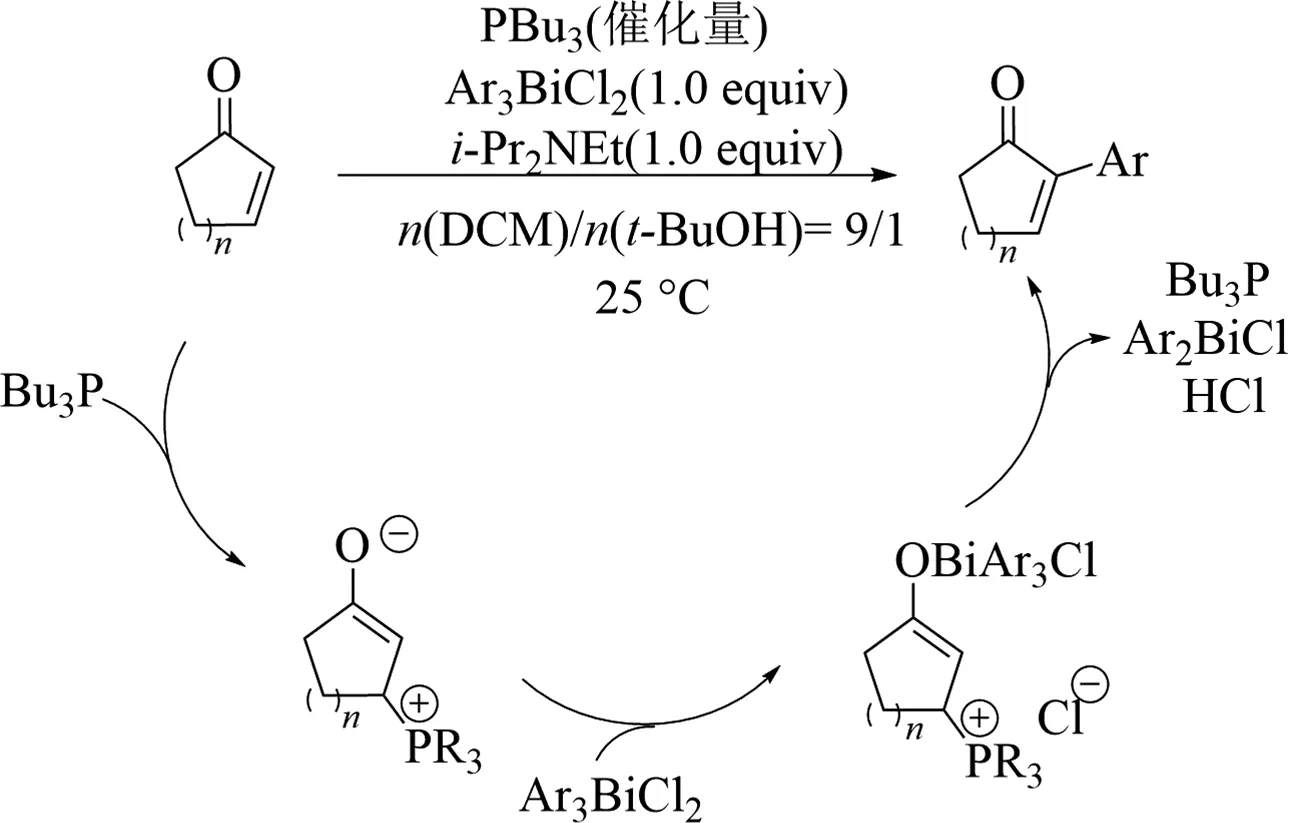
圖6 α,β-不飽和羰基化合物與芳基鉍試劑偶聯反應機理[28]
2.2 金屬催化偶聯
2.2.1α-芳基重氮化合物的偶聯反應
2008年,Peng等[29]報道了一種α-重氮羰基化合物與芳基硼酸的交叉偶聯反應.該反應使用鈀催化劑,α-重氮羰基化合物10與芳基硼酸11在含有二異丙胺及苯醌的甲苯溶液中進行反應,反應條件溫和,且反應速率快,80 ℃反應15 min即可得到82%的產率(12a)(見圖7).
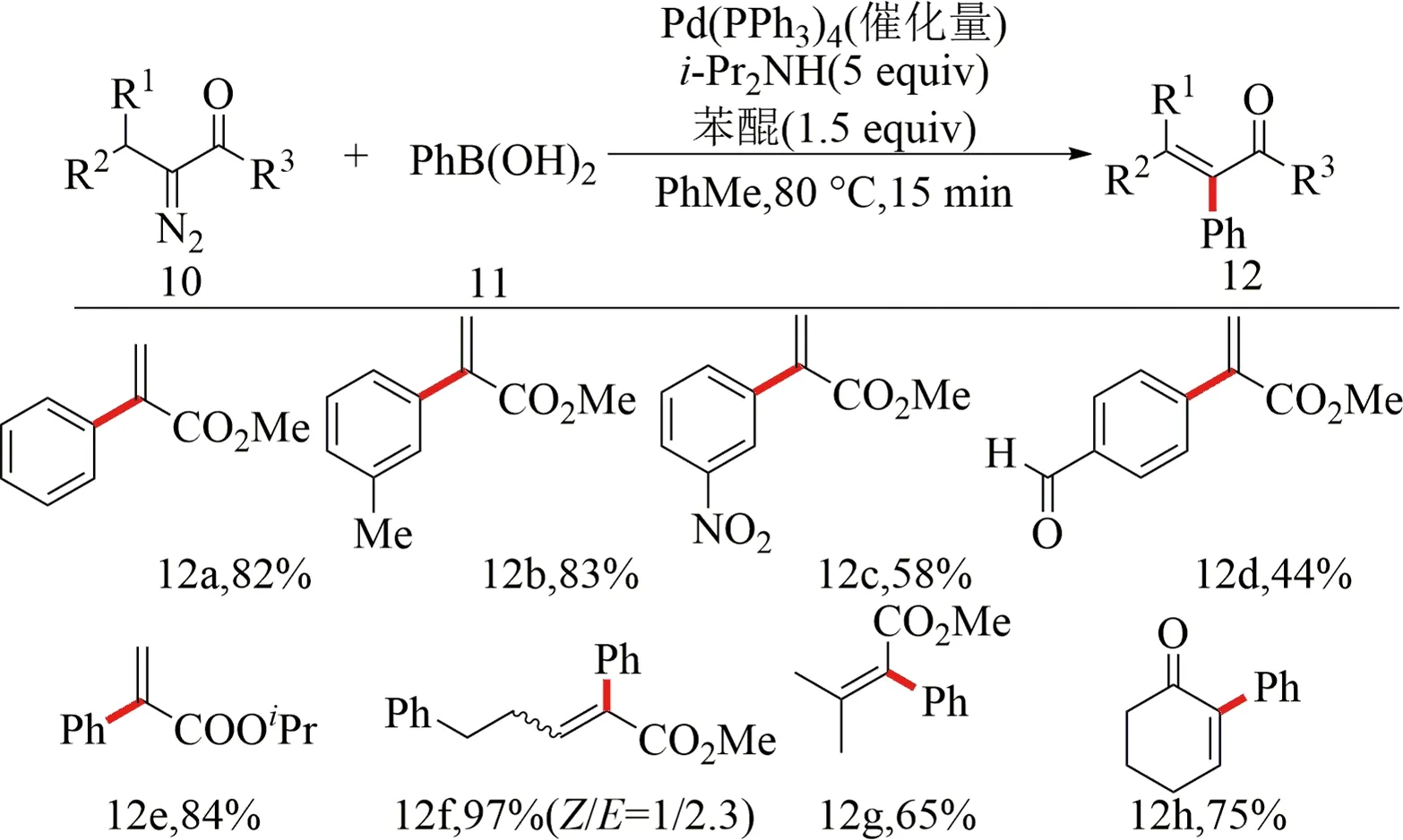
圖7 α-重氮羰基化合物與芳基硼酸的交叉偶聯反應[29]
2009年,Yu課題組[30]報道了在鈀催化劑作用下,芐基溴13與α-芳基重氮乙酸甲酯14的直接偶聯反應,合成了β-芳基-α-芳基丙烯酸甲酯15.該反應能選擇性合成E-丙烯酸甲酯,表現出較高的區域和立體選擇性(E/Z>30/1).與文獻[20]相比,反應產率明顯提高,且底物的范圍得到了進一步拓展,但部分產物產率低,如15d產率低于20%,15h產率僅8%(見圖8).
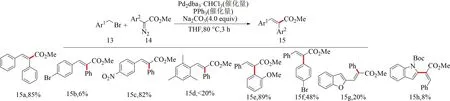
圖8 芐溴和α-芳基重氮乙酸甲酯催化偶聯反應[30]
2010年,Tsoi等[31]基于之前的工作報道了在鈀催化下,芳基硼酸和α-芐基重氮乙酸甲酯直接偶聯反應,該方法解決了Yu課題組[30]工作中15d產率低的問題,產率達到75%.
2.2.2 邁耶-舒斯特重排反應
邁耶-舒斯特重排反應是炔丙醇轉化成α,β-不飽和酮的一類重要反應,早在2009年Zhang等[32]便報道了基于邁耶-舒斯特重排反應構建α-芳基-α,β-不飽和羰基化合物的反應模式.該反應是乙酸炔丙酯16與芳基硼酸17在氟試劑(Selectfluor試劑)即1-氯甲基-4-氟-1,4-重氮化二環2.2.2辛烷雙(四氟硼酸)鹽及催化劑三苯基膦氯化金作用下實現均相氧化交叉偶聯反應,最終合成α-芳基-α,β-不飽和羰基化合物18,同時反應不可避免地存在化合物19及化合物20兩個副產物(見圖9).
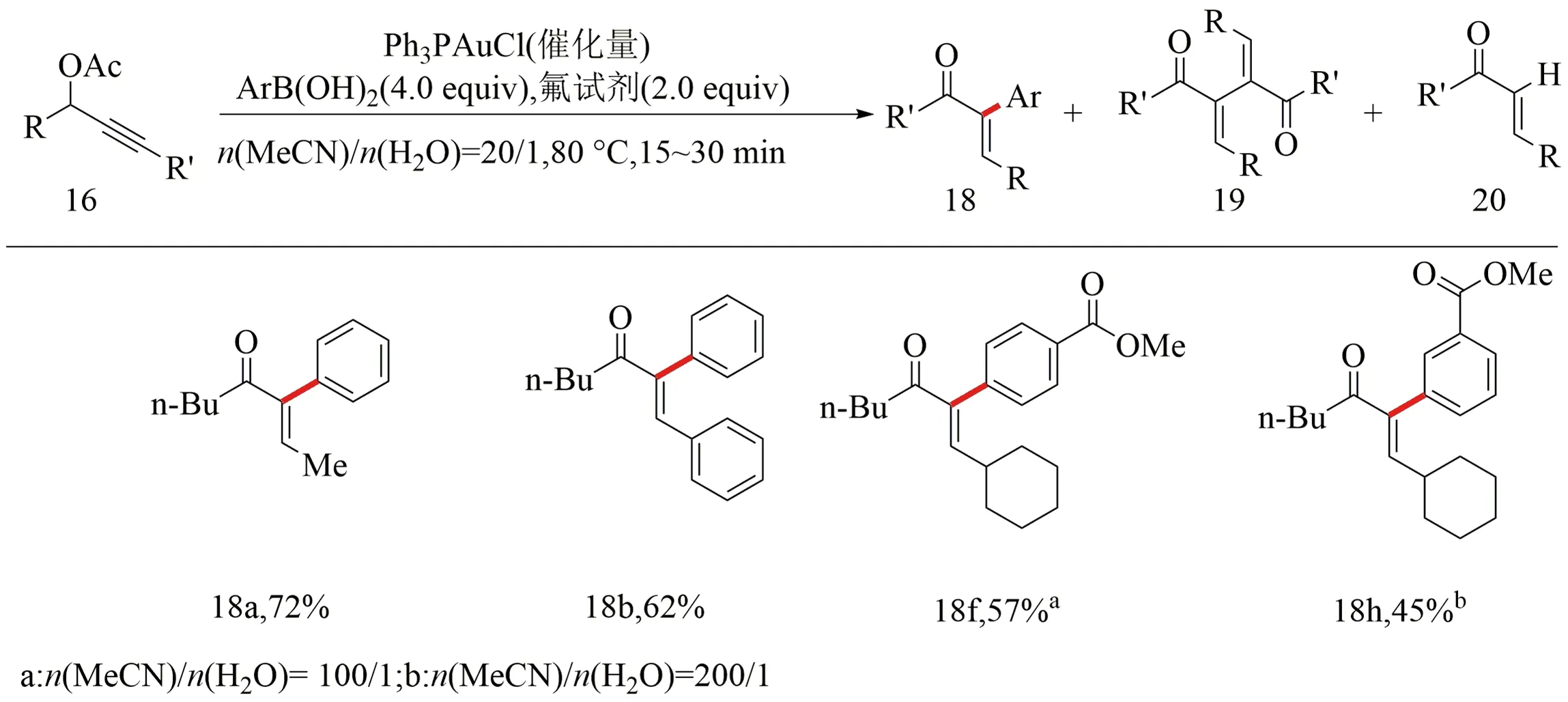
圖9 乙酸炔丙酯與芳基硼酸交叉偶聯反應[32]
Collins等[33]于2013年報道了炔丙醇與二芳基碘鎓鹽及DTBP(二叔丁基過氧化物)的邁爾-舒斯特重排反應,在氯化亞銅的催化作用下該反應只需要在二氯甲烷溶液中50 ℃回流3~6 h即可,該反應相對溫和,使用的催化劑氯化亞銅廉價易得.
在2016年以后,通過邁耶-舒斯特重排反應制備α-芳基-α,β-不飽和羰基化合物的反應模式得到了進一步的發展[34-37],例如Alcaide等[34]報道了使用可見光促進的炔丙醇與芳基重氮鹽的邁爾斯-舒斯特的芳基化反應.該反應在金催化劑和釕催化劑的雙重催化下,在室溫下使用可見光照射4 h即可反應完全.但該反應對于含給電子基團的芳基重氮鹽存在兼容性差的問題.
2.2.3 其他催化偶聯
2005年,Suginome等[38]報道了一類鈀催化的炔烴與氰基硼烷的加成反應,1-芳基-1-丙炔和氰基硼烷及鈀催化劑在二氧六環溶劑中130 ℃加熱12 h后得到β-硼烷-α-芳基-α,β-不飽和腈.
2012年,Samanta等[39]報道了由銠(Ⅲ)催化的色酮化合物21與烯烴化合物22直接氧化交叉偶聯反應,實現了色酮類化合物的α芳基化.但反應存在催化試劑昂貴及反應條件苛刻等問題,并且反應所報道的底物范圍較少(見圖10).
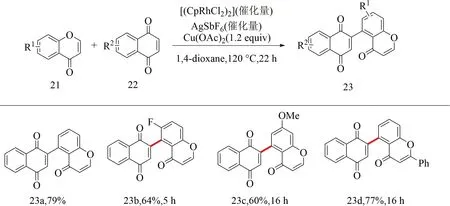
圖10 色酮化合物與烯烴化合物直接氧化交叉偶聯反應底物范圍[39]
2014年,Gandeepan等[40]在鈀催化劑的作用下,實現飽和羰基化合物與鹵代芳烴的偶聯反應,其中2-苯丙醛24與4-碘甲苯25在二甲基亞砜中于120 ℃反應15 h,得到芳基化產物(E)-2-苯基-3-(對甲苯)丙烯醛(26a),產率為85%.該反應對含吸電子基團的鹵代芳烴與含給電子基團的鹵代芳烴具有良好的官能團兼容性,如26c(76%),26d(83%).同樣對于酮類化合物,該反應模式也展現出良好的反應性.不足之處在于該反應僅對β位的芳基進行了底物范圍研究,未針對α位的芳基進行底物范圍研究(見圖11).
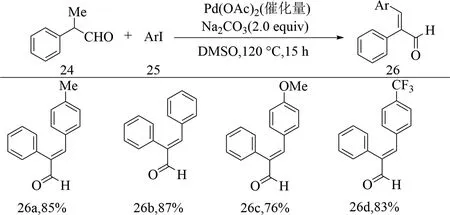
圖11 鹵代芳烴與飽和羰基的催化偶聯反應[40]
2020年,Yuen等[41]報道了鈀催化下異佛爾酮(3,5,5-三甲基-2-環已烯-1-酮)與氯苯的偶聯反應.
2.3 有機小分子催化
2011年,Song等[42]采用有機小分子催化策略,實現了α,β-不飽和的α芳基化.α,β-不飽和醛27與4-溴代苯酚28在含碳酸鈉的氯仿條件下,在有機胺類化合物催化下進行偶聯得到α芳基化產物29,與Banwell[9],Molander[27]反應相比,Song等使用更容易制備的有機胺催化劑,避免使用價格昂貴的金屬催化,且底物范圍得到了拓展,反應選擇性好,但產物29g產率僅為15%,產物29h產率為0%(見圖12).
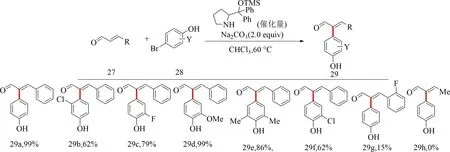
圖12 有機小分子催化合成α-芳基-α,β-不飽和醛的反應底物范圍[42]
Song等[42]認為,反應機理可能是α,β-不飽和醛27首先與有機胺類化合物反應生成亞胺離子,隨后4-溴苯酚在碳酸鈉的作用下與亞胺離子發生Friedel-Craft,得到中間體I,中間體I進一步發生環丙烷化得到中間體II,并在碳酸鈉的作用下,反應生成烯胺中間體III,隨后中間體III發生開環反應生成中間體IV,最后中間體IV脫除有機胺類化合物生成α-芳基-α,β-不飽和醛29(見圖13).
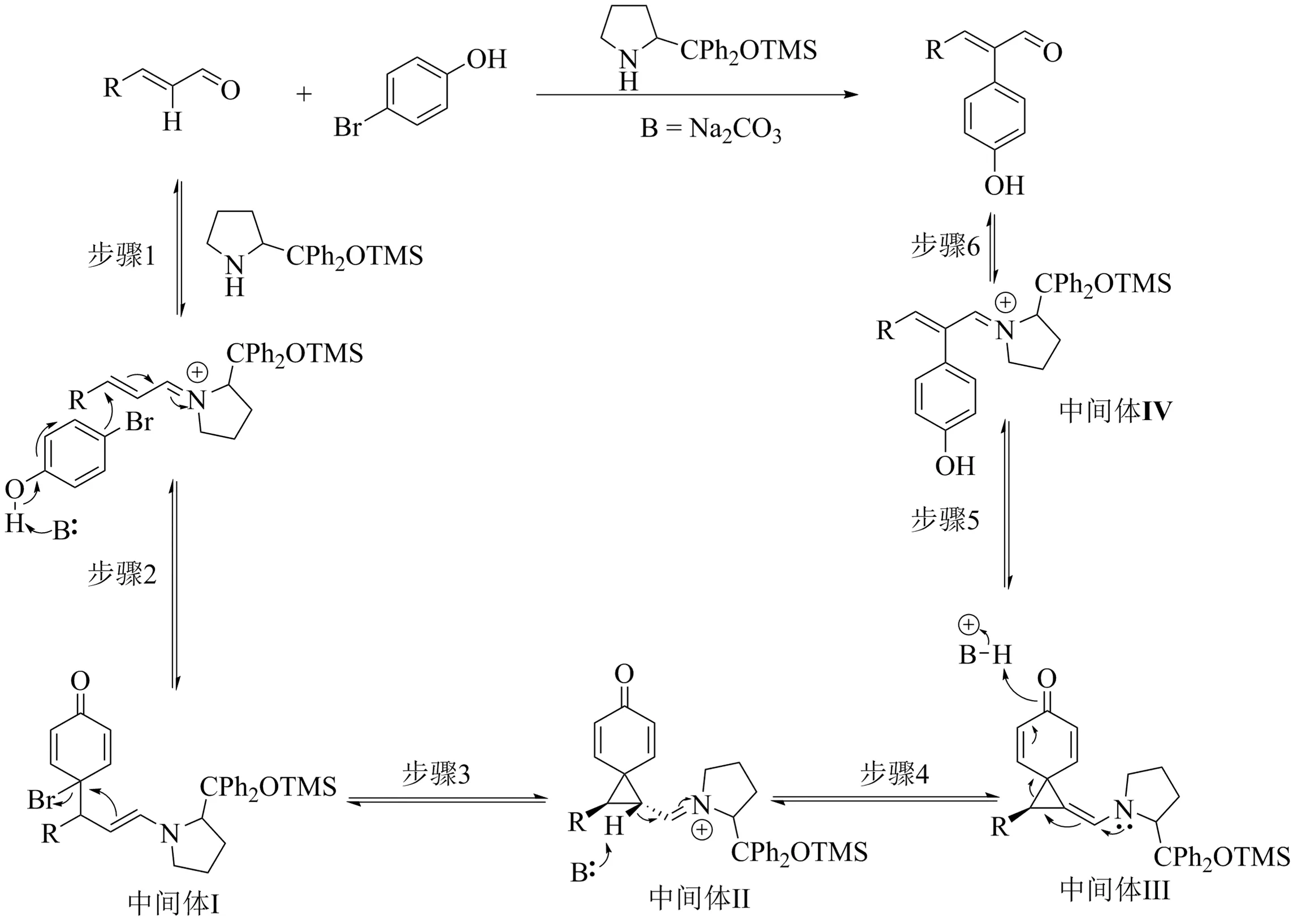
圖13 有機小分子催化合成α-芳基-α,β-不飽和醛的可能機理[42]
2.4 高價碘重排介導的芳基化
2020年,Sousa等[43]首次發展了利用高價碘重排反應實現α,β-不飽和羰基化合物的α芳基化.該反應首先通過2-環己烯酮30在三氟甲磺酸三甲基硅酯的作用下與吡啶加成原位生成β-吡啶基甲硅烷基烯醇醚31,隨后β-吡啶基甲硅烷基烯醇醚31與芳基高價碘試劑反應并發生[3,3]重排,最終制備得到α-芳基-α,β-不飽和羰基化合物32.該反應的高價碘試劑均可通過相應的文獻報道的方法進行合成[44-47],合成方法簡便.但該反應也存在局限性,產物32d,32e,32i產率低于20%(見圖14).
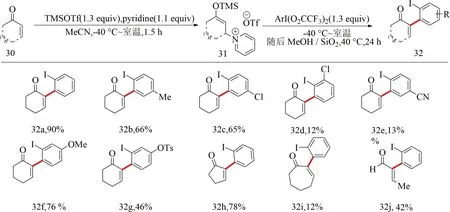
圖14 高價碘重排介導的芳基化反應[43]
2.5 芳基亞砜重排介導的芳基化
2020年,本課題組[48]首次發展了利用芳基亞砜與腈的[3,3]重排反應實現α,β-不飽和腈的α芳基化.該反應通過芳基亞砜33與α,β-不飽和腈34在三氟甲磺酸酐的活化作用下組裝形成乙烯基亞胺硫鎓鹽中間體.隨后,Lewis堿對該中間體進行Morita-Baylis-Hillman型加成,構建烯酮亞胺鹽型重排前體,重排得到MBH型重排產物,最后重排產物發生β-消除生成α-芳基α,β-不飽和腈35.該反應適用于大多數芳基亞砜和芳基烷基亞砜.但對于帶有給電子基團二芳基亞砜,則未能分離到預期產物35g(見圖15).
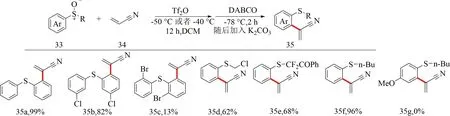
圖15 芳基亞砜與α,β-不飽和腈芳基化反應底物范圍[48]
3 結 論
在過去的幾十年中,α-芳基-α,β-不飽和羰基化合物的合成研究得到了一定的發展,提出了金屬催化偶聯、有機小分子催化偶聯及有機重排介導的合成方法.雖然一些合成方法存在底物范圍窄,反應條件苛刻,反應試劑昂貴等問題,但在對藥物研究需求不斷增加的未來,α-芳基-α,β-不飽和羰基化合物作為一類具有潛在藥用價值的有機官能片段將得到更多研究人員的關注.隨著有機合成方法學的不斷發展,更加綠色、高效并且環保的合成方法將被進一步開發.
