槲皮素的結構修飾及生物活性研究進展
2023-03-10 01:48:46李陽杰曹瑞梅毛雅君邵香敏馮亞莉翟廣玉
中草藥 2023年5期
李陽杰,曹瑞梅,毛雅君,邵香敏,馮亞莉,翟廣玉
鄭州工業應用技術學院藥學與化學工程學院,河南 鄭州 450041
槲皮素為廣泛存在于蔬菜(番茄、甘藍、蘆筍等)、水果(蘋果、杏、草莓等)、中藥(槐米、絞股藍、銀杏葉等)中的黃酮類化合物[1-2],具有抗氧化、抗炎、降糖、抗癌、預防和治療心腦血管疾病等藥理作用[3-10]。隨著科學技術的發展,人們逐漸認識到,從天然產物中獲得的化學成分應用于防病治病效果更好。因此,利用植物中的化學物質促進健康,已成為研究者廣泛關注的熱點[11]。
槲皮素是具有多種生物活性的天然抗氧化劑,可調節眾多與疾病進展有關的細胞內、外信號通路。越來越多的研究也表明通過合成新的槲皮素衍生物可以改善槲皮素較低的溶解度和生物利用度。為了使槲皮素發揮其預防和治療疾病的作用,盡快應用
于臨床,研究學者對槲皮素進行結構修飾,主要包括對羥基的修飾生成醚和酯,對羰基的修飾生成羰基氧被取代的產物,對槲皮素A、B 環的修飾等。通過優化修飾獲得了溶解性能好、生物利用度高、活性明顯改善、抗癌活性增強的槲皮素衍生物[12-14],如圖1所示。本文通過對槲皮素衍生物的結構修飾以及其抗癌、抗糖尿病、抗菌、抗炎、抗病毒等生物活性進行綜述,并對其構效關系進行分析,為天然產物的研究、開發及利用提供參考。
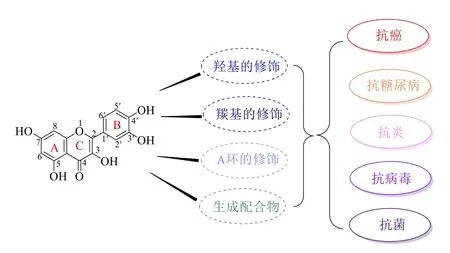
圖1 槲皮素的結構修飾及生物活性Fig.1 Structural modification of quercetin
1 槲皮素的結構修飾與生物活性
1.1 槲皮素抗癌衍生物的合成
1.1.1 羥基的結構修飾 前列腺癌是美國男性最常見的浸潤性癌癥,也是癌癥相關死亡的第2 大原因。雖然有幾種療法在治療早期取得了成功,但由于多杉醇一線治療的耐藥性問題,目前對晚期轉移性前列腺癌的治療仍然無效。Al-Jabban 等[15]采用水溶性四唑鹽(water-soluble tetrazole salt,WST-1)細胞增殖實驗檢測了18 個合成槲皮素衍生物對人前列腺癌LNCaP、DU145、PC-3 細胞的抗增殖作用,結果表明槲皮素中C-3、C-4?和C-7 羥基與短鏈烷基(如甲基或乙基)的烷基化略微增加了對3 種人前列腺癌細胞的抗增殖活性。然而,當支鏈烷基(1p 異丙基和1q 異戊基)或長鏈烷基(1j 丙基、1l丁基、1n 戊基、1o 己基)引入到這3 個位置時,其生物活性減弱。此外,4 種3,7-二烷基槲皮素(1b、1e、1i、1k)對3 種前列腺癌細胞的抑制率都明顯高于槲皮素,其半數抑制濃度(half inhibitory concentration,IC50)是槲皮素的2~11 倍。構效關系表明:①槲皮素的C-3、C-4?和C-7 羥基同時引入3 個或較長的烷基導致前列腺癌細胞的抗增殖活性顯著喪失;②在槲皮素的C-3 和C-7 羥基中,化合物1b 結合2 個甲基,1e 結合2 個乙基,1i 結合2個丙基,1k 結合丁基的衍生物前列腺癌細胞的抗增殖活性更強。即3,7-O-二烷基槲皮素比3,4?,7-O-三烷基槲皮素對前列腺癌細胞的抑制作用更強。
槲皮素溶于甲醇,加入無水K2CO3的N,N-二甲基甲酰胺(N,N-dimethylformamide,DMF)溶液,加入適當的鹵代烷;反應混合物在室溫下攪拌12~48 h,然后用乙醚-醋酸乙酯(1∶1)稀釋。用鹽水沖洗,有機層在無水硫酸鎂上干燥,濾過后在真空中濃縮,得到相應的粗品。對粗品進行柱色譜純化,用醋酸乙酯-正己烷(3∶7)作洗脫劑,得到化合物1a~1r,見圖2。
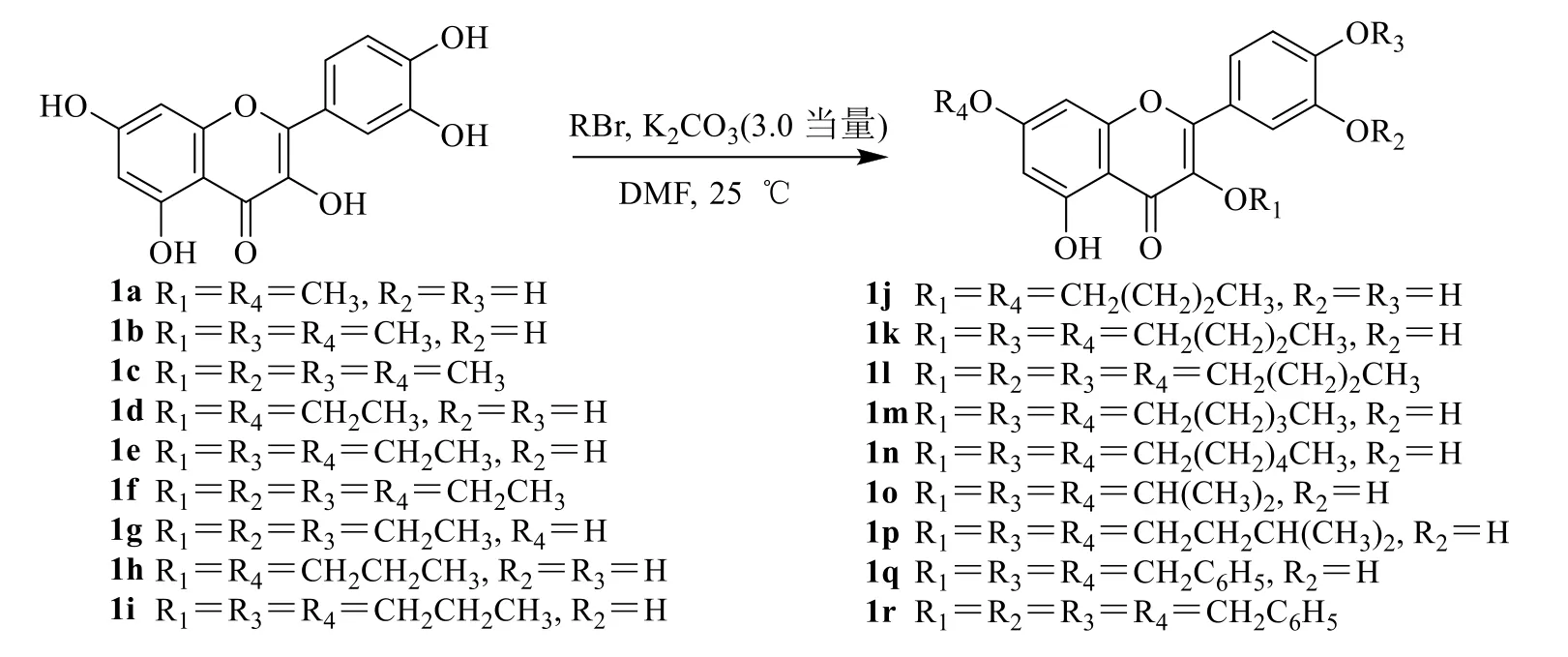
圖2 槲皮素烷基醚的合成Fig.2 Synthesis of quercetin alkyl ether
為了獲得具有良好細胞毒性的新型槲皮素衍生物,Bao 等[16]使用MTT 法測定評估2a~2f 和母體化合物槲皮素對人乳腺癌MCF-7 細胞和人結腸癌Caco-2 細胞的細胞毒性。結果顯示2a 和2f 對MCF-7 和Caco-2 細胞的IC50值低于槲皮素。進一步評估了2f 對人肺癌NCI-H446、A549 細胞、人胃癌MGC-803 和SGC-7901 細胞的細胞毒性,并與槲皮素進行了比較。結果表明2f 對這4 種癌細胞系均表現出強烈的細胞毒性,并且強于槲皮素,有望成為癌癥化療藥物。
槲皮素溶于吡啶,加入乙酸酐反應,在丙酮中重結晶得到五乙酰槲皮素。將五乙酰槲皮素溶于18-冠-6 和CH3CN 與鹵代烴(正丁基溴、烯丙基氯、肉桂酰氯、香葉基)反應,濾過,將濾液用石油醚萃取,濃縮,過色譜柱,以石油醚-醋酸乙酯(4∶6)洗脫,旋蒸,得到2a~2g,見圖3。

圖3 槲皮素-3,7-烷基醚的合成Fig.3 Synthesis of quercetin-3,7-alkyl ethers
為了通過化學修飾來設計具有更高效力的槲皮素衍生物,以達到治療前列腺癌的目的。Rajaram等[17]使用WST-1 法測定24 個O-四甲基槲皮素含氮衍生物對PC-3、DU-145 和LNCaP 細胞的體外抗增殖活性。以菲斯汀、槲皮素為陽性對照,二甲基亞砜為陰性對照,結果表明3a~3x 均比槲皮素和菲斯汀具有更高的抗增殖活性。其中,在DU145 細胞中,有15 個衍生物的活性明顯高于陽性對照藥,衍生物3j 和3k 被確定為2 個最佳衍生物,IC50值分別為1.73、6.53 μmol/L。N,N-二戊氨基是3-O-氨基烷基-3?,4?,5,7-O-四甲基槲皮素抗增殖活性的最佳含氮基團。同時,24 個衍生物在抑制PC-3 和LNCaP細胞增殖方面比抑制DU145 細胞增殖更有效。其中,3j 抑制PC-3 細胞的增殖活性是槲皮素的35~182 倍,可以顯著誘導PC-3 細胞凋亡,有望作為抗前列腺癌藥物進一步開發。
選擇3?,4?,5,7-O-四甲基槲皮素(3A)作為母體化合物,因為在3-OH 上連接烷基,可以在一定程度上克服槲皮素中酚羥基引起的藥動學限制。3A 是以蘆丁為原料,通過甲基化和糖苷水解合成。3A 通過與二溴烷烴的O-烷基化反應,得到3-O-溴代烷基-3?,4?,5,7-O-四甲基槲皮素(3B~3D),再與胺發生N-烷基化反應,得到24 個3-O-氨基烷基-3?,4?,5,7-O-四甲基槲皮素(3a~3x),見圖4。
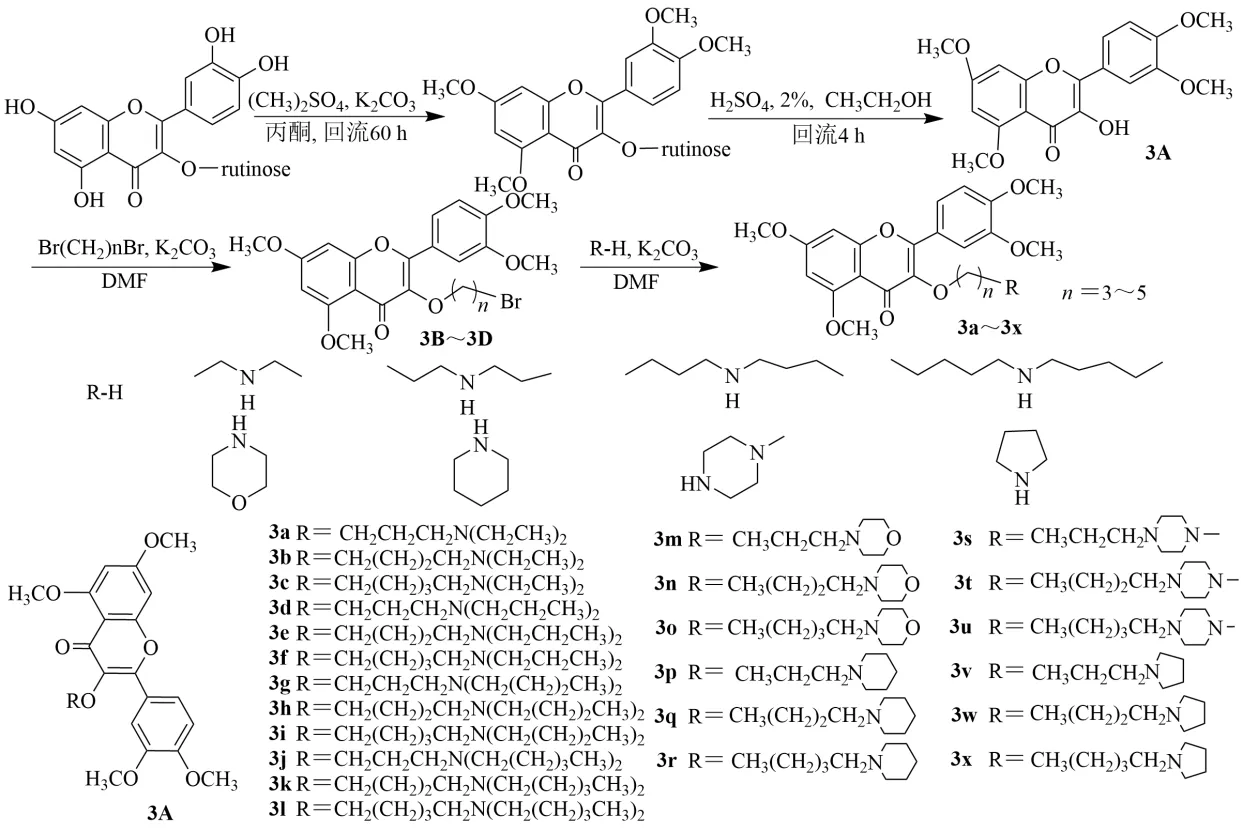
圖4 槲皮素四甲基烷基氨基醚的合成Fig.4 Synthesis of quercetin tetramethyl alkyl amino ether
Mukherjee 等[18]通過在槲皮素C-3?和C-5 位用不同的取代基修飾,考察10 個槲皮素衍生物脂溶性、水溶性和抗腫瘤活性。為了解決溶解性問題,在化合物4a 的C-3?位選擇性引入乙酸酯官能團,化合物4d 在人結直腸癌HCT116 細胞中的IC50值為3.03 μmol/L,在化合物4b 的C-5 位引入酯基,化合物4j 的IC50值為0.34 μmol/L。與槲皮素相比,在C-5 位引入酯基提高了4j 的溶解性(約為380倍)和細胞毒性(約為150 倍)。相應的酯水解物4e(IC50值為2.69 μmol/L)與其酯類4d 顯示出相似的活性,細胞毒性約為槲皮素(IC50值為45.32 μmol/L)的15 倍,4k 的活性比其酯衍生物4j 降低了4 倍,而4a 的二取代酯衍生物4g(IC50值為57.5 μmol/L)和它的水解衍生物4h(IC50值為7.34 μmol/L)都失去了活性,表明C-5 和C-3?的雙取代可能不是很理想,C-5 或C-3?與酯和相應的游離酸基團的單取代物其細胞毒性顯著增加。在C-5 和C-3?位引入含有弱堿基的脂肪族連接物,如N-甲基哌嗪和吡咯烷,單一取代得到4o、4p 和4v。與槲皮素相比,含吡咯烷的化合物4o 細胞毒性(IC50=1.98 μmol/L)增加22 倍,溶解度增加325 倍;含N-甲基哌嗪的化合物4p 中的C-3'位使其活性顯著升高,IC50值為0.48 μmol/L,增加了90 倍,同時溶解度增加約400倍。而4p 沒有顯示對外周血單個核細胞的毒性,表明4p 對癌細胞有選擇性毒性。4v 中C-5 位N-甲基哌嗪部分產生類似的效力,IC50值為0.55 μmol/L。
進一步研究顯示,與槲皮素相比,化合物4p 在HCT116 細胞中的細胞毒性增加了96 倍。此外,在CT-26 荷瘤小鼠實驗中表明,與槲皮素相比,4p 可增加荷瘤小鼠的存活率并使腫瘤體積減小(60%),說明槲皮素衍生物應用于臨床具有較大的潛力。
槲皮素、溴化芐、K2CO3在DMF 溶液中反應,通過柱色譜分離出4a 和4b。4a 與1.5 當量的溴乙酸甲酯反應,得到C-3?位的單乙酸酯取代物4c。4a與3.0 當量的溴乙酸甲酯反應,得到二取代物4f。化合物4c 用氫氧化鈀脫芐得到化合物4d,堿作用下水解得到化合物4e。化合物4f 脫芐后得到4g,酯水解得到化合物4h。4b 與1.5 當量的溴乙酸甲酯反應,得到C-5 位的單取代物4i。4i 脫芐后得到4j,酯水解得到化合物4k。用1.5 當量的1-溴-3-氯丙烷處理4a,轉化為4l,與吡咯烷和N-甲基哌嗪反應得到化合物4m 和4n。去芐基得到化合物4o 和4p。4a 用過量的1-溴-3-氯丙烷處理,轉化為4q,與吡咯烷反應得到化合物4r。去芐基得到二取代產物4s。4b 用1-溴-3-氯丙烷處理,轉化為4t,與N-甲基哌嗪反應得到化合物4u,去芐基得到化合物4v,見圖5。
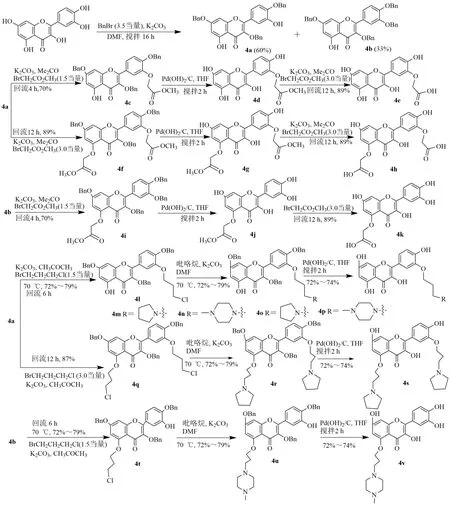
圖5 槲皮素-5-或-3′-醚基乙酸衍生物的合成Fig.5 Synthesis of quercetin-5 or-3′-ether acetic acid derivatives
由于對氧化應激的不穩定性,槲皮素在細胞培養液中分解,細胞中的槲皮素濃度低于最初添加的濃度。Kim 等[19-20]將其代謝和化學敏感的羥基7-OH和3-OH 分別用新戊氧甲基(pivaloxymethyl,POM)暫時封閉,得到2 種新的槲皮素衍生物5a 和5b。其中5a 孵育后的半衰期延長至4 h,通過共聚焦活細胞成像和高效液相色譜分析表明,5a 能有效地被細胞攝取,5a 及其水解產物在細胞內的濃度可維持12 h,優于槲皮素。同時,5a 能有效被細胞攝取,其穩定性和胞內積累表現為不依賴于穩定劑的細胞抑制作用和誘導細胞凋亡。5b 比5a 更穩定,但它不能穿透細胞膜。
乙酸酐加入到槲皮素的吡啶溶液中反應得到白黃色粉末5c。硫代苯酚緩慢加入到5c 和咪唑的N-甲基吡咯烷酮(N-methyl pyrrolidone,NMP)溶液中反應得到白色粉末5d。碳酸鉀和POM-碘(POM-I)添加到5d 的丙酮溶液中反應,通過硅膠柱色譜純化(己烷-醋酸乙酯1∶1)得到黃色粉末5a。
槲皮素與二氯二苯基甲烷反應得到5e,在吡啶中與過量的Ac2O 反應得到5f。用硫酚(PhSH)、NMP 和咪唑混合物選擇性除去化合物5f 7-O 位的乙酰基基團得到5g,再用芐基重新保護,得到關鍵中間體5h。為了選擇性地在3-O 位引入POM 促進劑,用甲醇氨處理去除乙酰基,在槲皮素的C-3 和C-5 得到具有游離酚羥基的5i。羰基氧和5-OH 之間的分子內氫鍵阻止了5-OH 的化學反應,這導致POM-I處理時5i 在3-O 位的區域選擇性烷基化。在氫解條件下,二苯甲縮酮和芐基同時脫保護,以67%的產率得到所需的5b,見圖6。
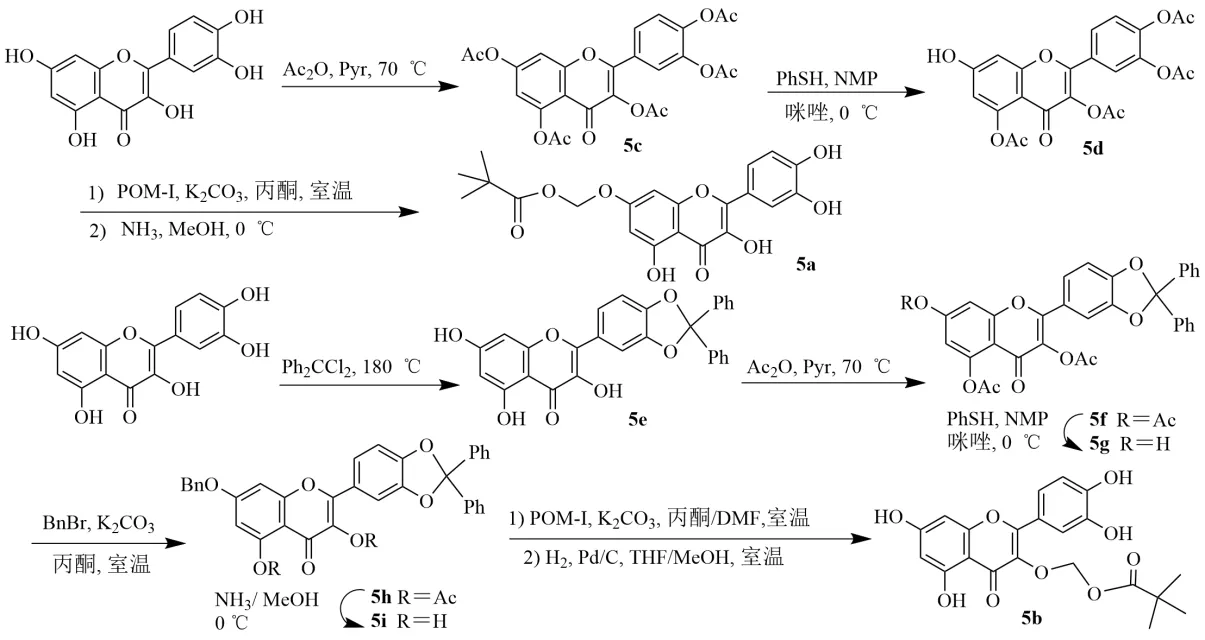
圖6 槲皮素-3-或-7-O-新戊氧甲基醚的合成Fig.6 Synthesis of quercetin-3-or-7-O-neopentyloxymethyl ether
Yuan 等[21]合成了15 種槲皮素衍生物,并評價其對腫瘤細胞株糖蛋白(P-glycoprotein,P-gp)、乳腺癌耐藥蛋白(breast cancer resistance protein,BCRP)和多藥耐藥相關蛋白1(multidrug resistancerelated protein 1,MRP1)的調節活性,結果表明合成的槲皮素衍生物具有良好的P-gp 和BCRP 介導的多藥耐藥逆轉活性。末端苯環上的甲氧基數目和O-3 側鏈上的連接基類型是決定槲皮素衍生物P-gp調節活性的2 個關鍵結構特征。化合物6l 具有較高的P-gp 調節活性。化合物6d、6i、6l 顯示出良好的BCRP 調節活性。此外,化合物6l 對P-gp 和BCRP具有等效性。由于其雙重調節活性,可能是一種很好的多藥耐藥性(multidrug resistance,MDR)逆轉劑。以上研究表明,槲皮素衍生物可作為P-gp 或BCRP 介導的癌細胞耐藥安全有效的調節劑。
蘆丁與甲基碘反應生成四甲基蘆丁(6a),酸性水解得到6b。6b 在K2CO3存在下與2-溴乙醇回流得到化合物6c。在K2CO3存在下,槲皮素與甲基碘在DMF 中反應,得到化合物6d 和6e。6b 與3,4,5-三甲氧基芐基甲磺酸酯生成化合物6g,與2-溴-1-(3,4-二甲氧基苯基)乙酮反應得到化合物6h,與3,4,5-三甲氧基苯甲酸反應得化合物6f。中間體6c與(E)-3-(3,4,5-三甲氧基苯基)丙烯酸、3,4,5-三甲氧基苯甲酸、3,4-二甲氧基苯甲酸或4-甲氧基苯甲酸進行酯化反應,得到化合物6i~6l。為了研究含雜環的槲皮素衍生物,制備了3 個新化合物,即化合物6b 與9-(2-溴乙基)-6-氨基嘌呤、9-(2-溴乙基)-1,3-二甲基-2,6-羰基嘌呤或6-(2-溴乙氧基)-4-(3-氯-4-氟苯氨基)-7-甲氧基-喹唑啉反應,分別得到化合物6m~6o,見圖7。
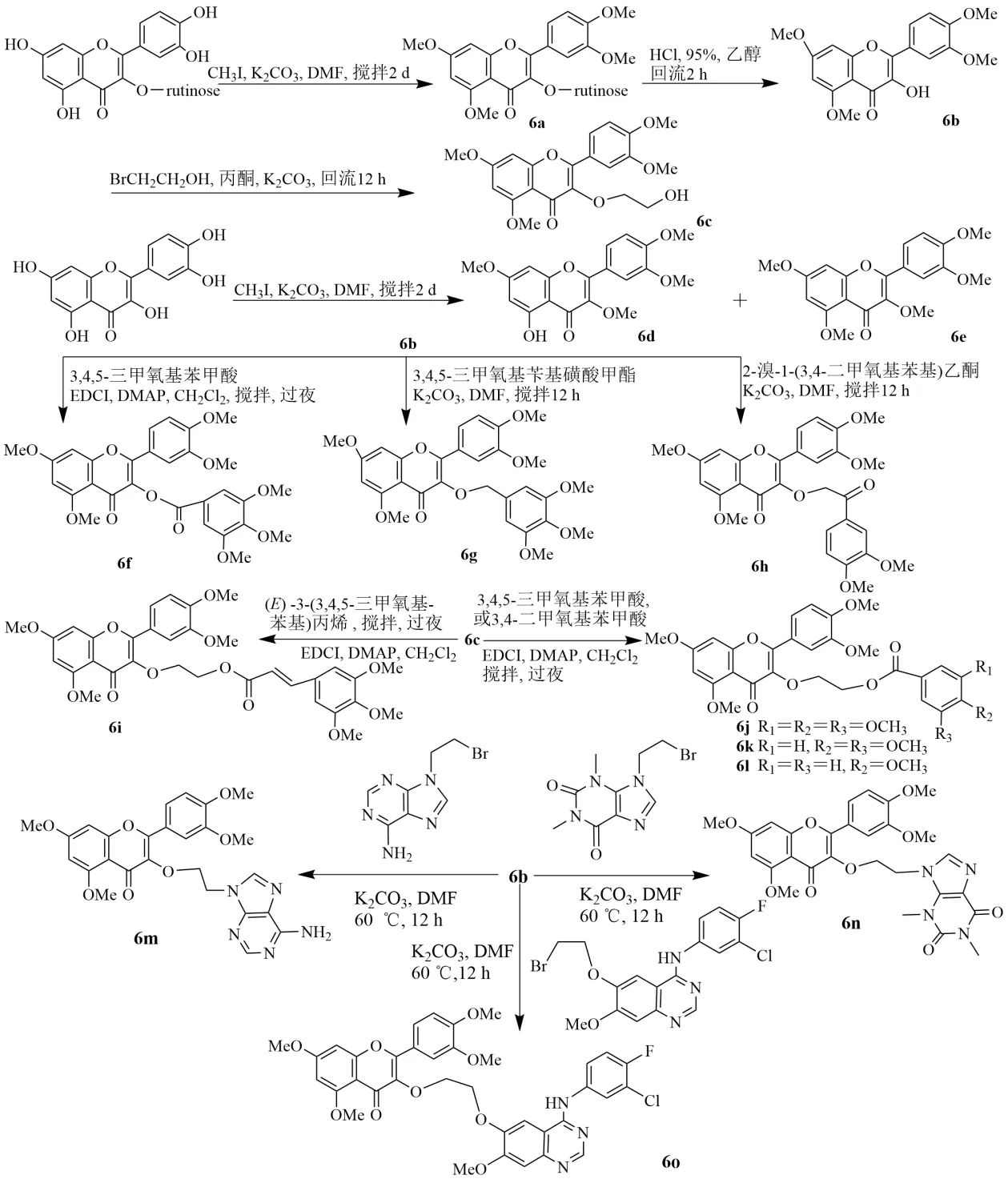
圖7 槲皮素-3-乙基醚衍生物的合成Fig.7 Synthesis of quercetin-3-ethyl ether derivatives
為了給篩選靶向表皮生長因子受體(epidermal growth factor receptor,EGFR)的新藥提供依據,Huang 等[22]合成了15 個槲皮素-3-O-氨基酸酯,采用酶聯免疫吸附法測定它們對EGFR 和酪氨酸蛋白激酶(sarcoma,Src)的抑制活性,結果表明槲皮素-3-O-氨基酸酯對EGFR 激酶的抑制率<43%,而對Src激酶的抑制率高達76%。表明槲皮素-3-O-氨基酸酯對Src 激酶抑制活性比EGFR 激酶更高。因此,在槲皮素中引入氨基酸基團可以逆轉EGFR 對Src 激酶的高選擇性抑制作用。為了證實其生物活性,進一步測定了化合物7a~7c、7g、7k 和7m 對Src 激酶的IC50值大于50%,結果表明,6 個化合物均具有顯著的抑制活性,IC50值為3.2~9.9 μmol/L。因此,為進一步開發以Src 激酶為靶點的新型抗癌藥物提供了一個有前景的新型結構。
蘆丁和K2CO3溶解在DMF 中,攪拌。加入芐基溴,在60 ℃攪拌,將混合物用10%醋酸調節pH為5,收集沉淀。沉淀物加到乙醇和濃鹽酸溶液中,在70 ℃攪拌2 h。混合物冷卻至室溫,沉淀濾過并用水洗滌。粗產物用二氯甲烷-乙醇重結晶得到7。N,N-二環己基碳二亞胺(dicyclohexylcarbodiimide,DCC)、4-二甲氨基吡啶(4-dimethylaminopyridine,DMAP)、叔丁氧羰基亮氨酸、化合物7 加入四氫呋喃(tetrahydrofuran,THF)溶液中,在室溫下攪拌。濾過混合物,濾液通過快速柱色譜(二氯甲烷-甲醇)純化得到7A,加入乙醇-二惡烷混合液,10%鈀碳,混合物在H2氣氛中,室溫下攪拌3 h。所得混合物通過硅藻土濾過,用乙醇洗滌并通過快速柱色譜純化(二氯甲烷-甲醇),得7a。使用與7a 相同的方法,從叔丁氧羰基丙氨酸(tertbutoxycarbonylalanine-OH,Boc-Ala-OH)得到7b。相同的方法可以得到7c~7n,見圖8。
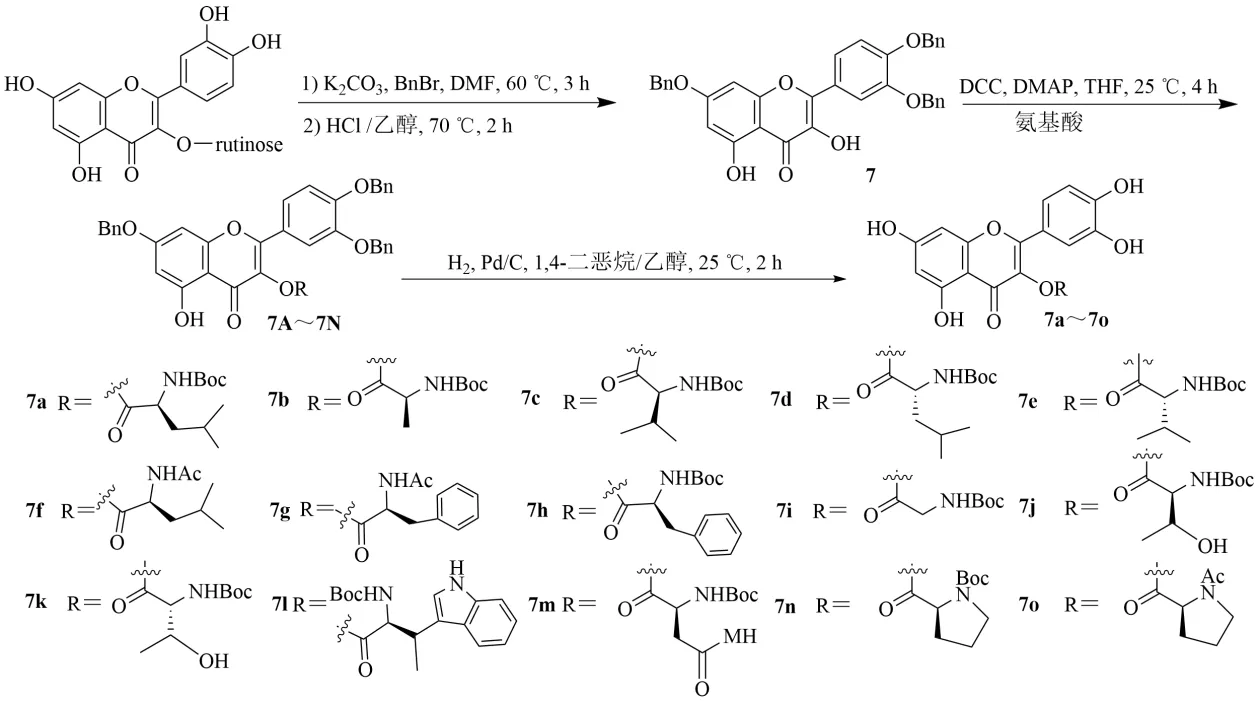
圖8 槲皮素-3-O-氨基酸衍生物的合成Fig.8 Synthesis of quercetin-3-O-amino acid derivatives
Kim 等[23]采用MTT 法測定槲皮素3-O 或7-O位的丙氨酸或谷氨酸衍生物(8a~8f)對HCT116 細胞、LNCaP 細胞和人包皮成纖維HS27 細胞的細胞毒性,結果表明,8a 和8b 顯示出最強的逆轉MDR活性(IC50=0.41、0.14 μmol/L),分別為槲皮素的20.0和58.6 倍。8c 和8d 顯示出中等的調節活性(IC50=1.21、1.10 μmol/L),與槲皮素(1.92 μmol/L)相比,略有增加。8e 和8f 的MDR 逆轉活性(IC50=0.78、0.71 μmol/L)分別為槲皮素的10.5、11.5 倍。8b 是逆轉人子宮肉瘤MES-SA/Dx5 細胞對阿霉素耐藥性最有效的藥物,也能增強同一耐藥細胞系中其他抗癌藥的細胞毒作用,其半數有效濃度(half effective concentration,EC50)為0.8~0.9 μmol/L。8b 可抑制P-gp 的藥物外排,P-gp ATP 酶分析表明8b 可與P-gp 的藥物結合位點相互作用,刺激其ATP 酶活性。對8b 的理化分析表明,谷氨酸的引入可顯著改善槲皮素的溶解性、穩定性和細胞攝取,使槲皮素氨基酸結合物成為安全的MDR 調節劑。
為了區域選擇性地將氨基酸引入槲皮素的結構中,需要對其5 個羥基進行選擇性保護。首先,槲皮素在180 ℃下與二氯二苯甲烷反應得到相應的二苯甲基酮,其余3 個羥基在吡啶中與過量的乙酸酐乙酰化,得到化合物8g。8g 在NMP 中與PhSH和咪唑反應得到7-O-單脫乙酰化產物8h。由于在堿性條件下乙酰基不穩定。因此,8h 的3,5-二乙酰基被替換為堿穩定的醚保護基團。通過順序保護-去保護策略,用甲氧甲基(methoxymethyl,MOM)保護8h 的游離7-OH,得到關鍵中間體8i。將8i 的3-OH基團保護為芐基醚,然后在酸性條件下裂解7-甲氧基醚鍵,得到8j,再與8o 或8p 偶聯,得到相應的槲皮素7-氨基甲酸酯。二苯甲基縮酮和芐基保護基同時氫解,然后使用三氟乙酸(trifluoroacetic acid,TFA)促進叔丁酯的裂解,得到8a 和8b。將8i 與(S)-N-Boc-Ala 或(S)-N-Boc-Glu(OtBu)進行1-乙基-3-[3-(二甲氨基)丙基]-碳二亞胺偶聯,然后去除7-OMOM 官能團(8k),得到8l。去掉8k 和8l 的二苯基甲縮醛和叔丁酯,分別得到游離的槲皮素3-酯(8c和8d),N-Boc 保護的8k 或8j 與8o 或8p 進一步偶聯,隨后的去保護反應得到槲皮素7-氨基甲酰基-3-酯(8e 和8f),見圖9。

圖9 槲皮素-3-O-或-7-O-氨基酸酯的合成Fig.9 Synthesis of quercetin-3-O-or-7-O-amino acid ester
Kellici 等[24-25]使用MTT 法測定槲皮素-3?或-4?-O-氨基酸酯類物質對DU-145、PC-3 細胞的細胞抑制活性和細胞毒活性,結果表明槲皮素-谷氨酸(9g、9o)和槲皮素-丙氨酸(9e、9m)是最有效的結合物,具有較高的細胞抑制活性。槲皮素-亮氨酸(9f、9n)和槲皮素-苯丙氨酸(9h、9p)細胞抑制活性較低。
氨基酸在密閉容器中與高氯酸和乙酸叔丁酯反應生成氨基酸叔丁酯。使用雙(4-硝基苯基)碳酸酯和N,N-二異丙基乙胺(N,N-diisopropylethylamine,DIPEA)在THF 中將氨基酸叔丁酯轉化為活性氨基酸甲酸酯,然后用槲皮素進行醇解。通過非水水解(TFA/二氯甲烷)實現了叔丁基保護基團的最終脫保護,得到槲皮素氨基酸酯。
將DIPEA 添加到氨基酸叔丁酯的溶液中,加入雙(4-硝基苯基)碳酸酯、THF,在室溫氮氣環境下,攪拌12 h,加入槲皮素并攪拌反應,混合物在室溫下再放置12 h。經薄層層析驗證,直到槲皮素完全消失。旋蒸,粗品柱色譜,二氯甲烷-甲醇(8∶2)洗脫,洗脫液旋蒸得黃色固體化合物(9a~9d 和9i~9l)。加入二氯甲烷、TFA,在0 ℃下攪拌4 h。反應完成后,加入己烷,濾過,得黃色沉淀,用己烷洗2 次,加入二氯甲烷溶解,柱色譜,二氯甲烷-甲醇(7∶3)洗脫,旋蒸得黃色固體化合物(9e~9h和9m~9p)。所的化合物用核磁、質譜和紅外光譜進行表征,可得到主要的異構體3'-O-取代的槲皮素(9a~9d、9e~9h)以及次要的異構體4'-O-取代的槲皮素(9i~9l、9m~9p),見圖10。
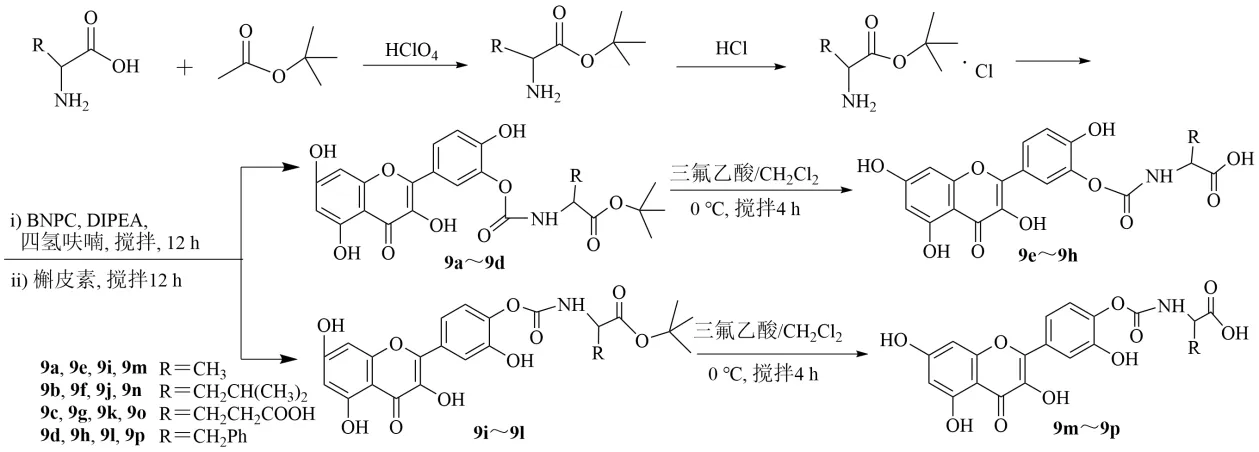
圖10 槲皮素-3′-或-4′-O-氨基酸酯的合成Fig.10 Synthesis of quercetin-3′-or-4′-O-amino acid esters
Lo 等[26]評估了槲皮素-5-O-酰基酯(10a~10g)對HCT116 細胞和人乳腺癌MDA-MB-231 細胞的抗增殖活性,及其針對1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picryl-hydrazyl radical,DPPH)自由基清除活性,結果表明10d、10e 和10g 具有較好的活性,其中衍生物10e 對受試癌細胞的IC50值最低,分別為(1.53±0.02)、(1.51±0.01)μmol/L,對自由基清除活性沒有改變。通過比較發現長酰基衍生物比短酰基衍生物具有更好的整體活性,表明親脂性的增加會導致生物利用度和活性的提高。一般來說,具有長酰基槲皮素衍生物比短酰基槲皮素衍生物顯示出更好的抗HCT116 和MDA-MB-231 細胞的活性。5-O-酰基碳鏈較長的衍生物對DPPH 自由基的清除活性與槲皮素相當,而酰基碳鏈較短的衍生物活性略有降低。
槲皮素溶于二苯醚中,加入二苯二氯甲烷,攪拌,加入石油醚,沉淀粗產物。濾過沉淀,溶解在EtOAc 中,真空除去溶劑。所得粗產物通過快速色譜法(石油醚-EtOAc 4∶1)得到黃色固體狀化合物10h。10h 和K2CO3在DMF 中攪拌溶解,室溫下加入溴化芐,攪拌,所得混合物用二氯甲烷萃取。合并萃取液,真空除去溶劑。粗產物經純化快速色譜法(石油醚-EtOAc 9∶1)得到黃色固體化合物10i。將10i 溶于三乙胺和二氯甲烷中,攪拌,加入乙酰氯,攪拌,用二氯甲烷萃取,合并有機層,真空除去溶劑,閃蒸提純色譜得到白色固體化合物10A。相同方法可得到10B~10G。10A 和20% Pd(OH)2/C,攪拌,快速色譜法(石油醚-EtOAc 1∶1)純化,得到黃色固體狀化合物10a。相同方法可得到10b~10g,見圖11。

圖11 槲皮素-5-酰基酯的合成Fig.11 Synthesis of quercetin-5-acyl ester
1.1.2 羰基的結構修飾 MDA-MB-231 細胞與其他類型的乳腺癌細胞不同,由于缺乏雌激素、孕激素以及EGFR2 的激素受體,故其不適合激素治療。Adnan 等[27]使用MTT 法測定了6 個槲皮素衍生物抗腫瘤細胞增殖活性,實驗顯示,化合物11a 和11d都使細胞活力顯著降低,分別降至43.7%和38.1%(其他化合物分別為11b 68.6%,11c 58.0%,11e、11f 無降低)。化合物11a 和11d 的IC50值分別為2.042、1.838 μmol/L,表明其具有針對三陰性乳腺癌類型的潛在抗癌活性。
槲皮素加入丙酮、K2CO3,攪拌,加入硫酸二甲酯,加熱回流,濾過,熱丙酮洗3 次,濾液旋蒸,甲醇重結晶,得到化合物11a。11a 溶于甲苯,加熱至110 ℃,加入Lawesson 試劑 [2,4-雙(4-甲氧基苯基)-1,3-二噻二膦-2,4-二硫化物],回流,將反應混合物冷卻至室溫,濾過,沉淀二氯甲烷溶解,柱色譜,氯仿洗脫得到化合物11b。11b 溶于乙醇中,滴加水合肼,攪拌至深綠色溶液變為橙黃色。加入胺后5 min 內通入硫化氫。0.5 h 后,停止通入硫化氫,冷水加入到反應混合物中并放于冰箱中冷藏24 h。濾過,洗滌得化合物11c。室溫下11b 與過量的碘甲烷反應,反應混合物顏色從深綠色變為紅色,沉淀從溶液中結晶出來,24 h 后收集,乙醚洗滌得到化合物11d。苯胺與11d 在熱乙醇中反應,15 min 加入甲硫醇,產物通過硅膠柱分離,二氯甲烷-甲醇9∶1 洗脫,收集洗脫液,旋蒸得到化合物11e。將11d 懸浮在溫甲醇中,加入3,4,5-三甲氧基苯胺的甲醇溶液,溶液的紅色消失,反應3 h 產生黃色沉淀,沉淀物用氯仿洗滌,得到化合物11f,見圖12。
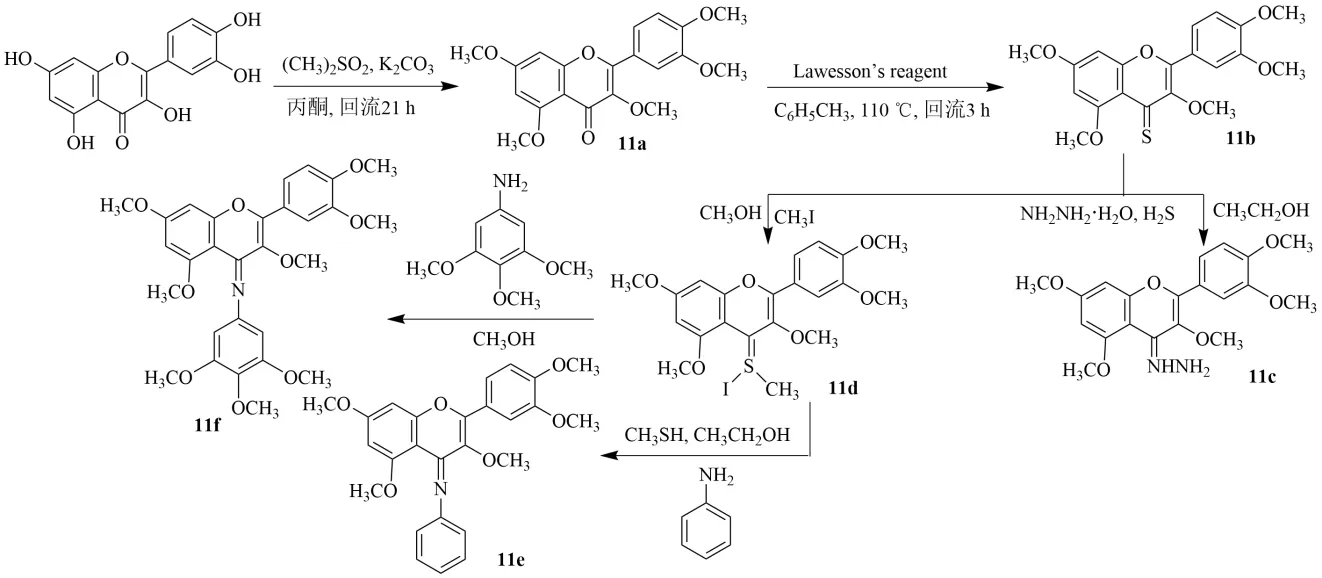
圖12 槲皮素羰基衍生物的合成Fig.12 Synthesis of quercetin carbonyl derivatives
Martins 等[28]應用MTT 法測定了3 個槲皮素硫或硒羰基衍生物對腫瘤細胞的生長抑制作用。對人結直腸腺癌HCT-15 細胞、MCF-7 細胞、多藥耐藥MCF-7-ADR 細胞、人宮頸腺癌A431 細胞活性進行測試,結果表明硒羰基衍生物12c 的細胞毒性高于其他硫族衍生物 [對于MCF-7 細胞,IC50值分別是12a>100 μmol/L,12c(3.08±1.98)μmol/L,12b、12d 無],充分證明硒羰基在細胞毒活性中的關鍵作用。對12c 與MCF-7 細胞的細胞毒性機制的初步研究表明,它們具有有效抑制克隆擴增的能力以及抑制硫氧蛋白還原酶活性導致細胞凋亡。與它們的羰基和硫代類似物相比,12c 顯示出更高的細胞毒性,還具有克服MDR 的能力。此外,12c 對惡性腫瘤細胞具有抑制作用,而對其他細胞無毒性。與硫屬元素衍生物和順鉑相比,它們表現出更高的選擇性。因此,12c 可作為開發潛在的癌癥化療候選化合物。
槲皮素與硫酸二甲酯反應,得到化合物12a。12a 與五硫化二磷在THF 中反應,室溫放置3 d,得到化合物12b。將12b 放入含有過量三溴化硼的二氯甲烷溶液中,室溫下反應,脫去保護基,得到化合物12d。為了制備硒衍生物,使用微波,2,4-雙(苯基)-1,3-二硒二膦烷-2,4-二硒化物(Woollins’試劑,WR)作為硒源[29],有效把羰基轉化為硒羰基。槲皮素、乙腈、WR 在175 W 微波輻照5 min 條件下得到化合物12c。然而,盡管成功地完成了硫衍生物12b 的脫保護,得到了12d,硒衍生物12c 脫保護沒有得到12e,見圖13。
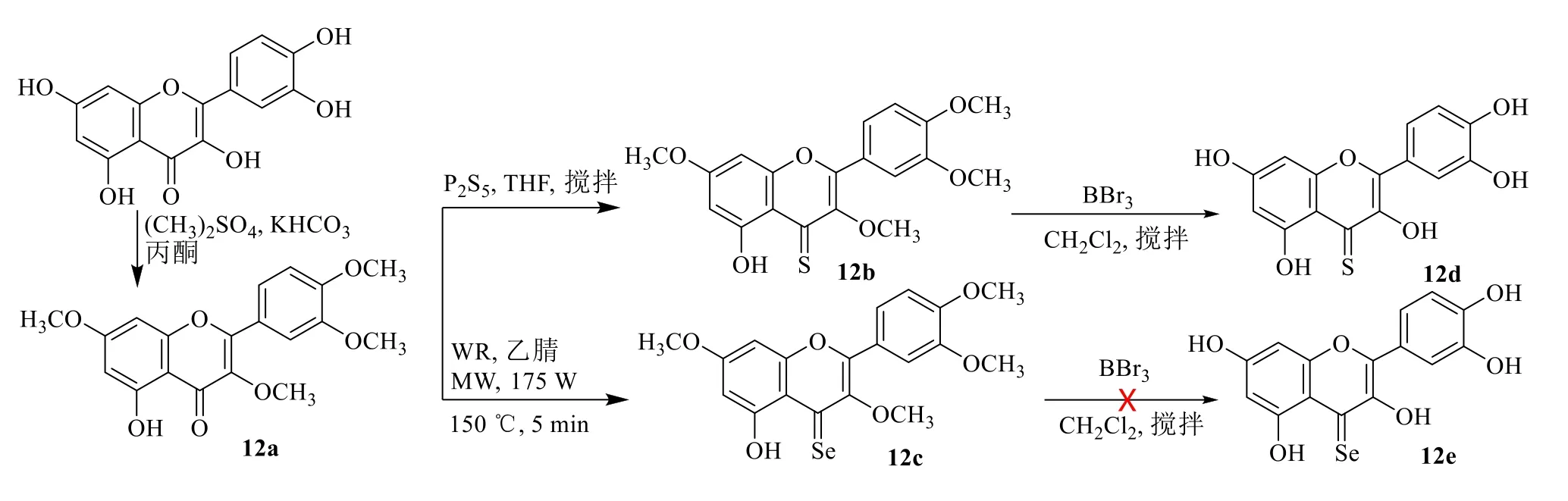
圖13 槲皮素硫或硒羰基衍生物的合成Fig.13 Synthesis of quercetin sulfur or selenium carbonyl derivatives
1.2 槲皮素抗糖尿病衍生物的合成
金屬及其配合物的藥用和臨床上的應用具有重要意義。Refat 等[30]通過注射鏈脲佐菌素誘導大鼠糖尿病模型,探討槲皮素鋅(II)(13a)配合物的抗糖尿病作用,結果表明13a 和骨髓間充質干細胞(mesenchymal stem cells,MSCs)聯用可顯著改善胰島素分泌,減少細胞炎癥,并有助于改善胰腺和糖代謝并發癥,比單獨使用MSCs 或13a 時效果更好。結果證實13a 對治療高血糖、遺傳毒性非常有效,為治療糖尿病及其相關并發癥提供了新方向。 向溶解在甲醇中的槲皮素中加入Zn(NO3)2·6H2O,加入氨水調pH 為8,室溫下攪拌4 h。濾過所得黃色產物室溫下緩慢蒸發過夜,然后用少量MeOH 洗滌,在干燥器中用無水CaCl2干燥得到13a,見圖14。
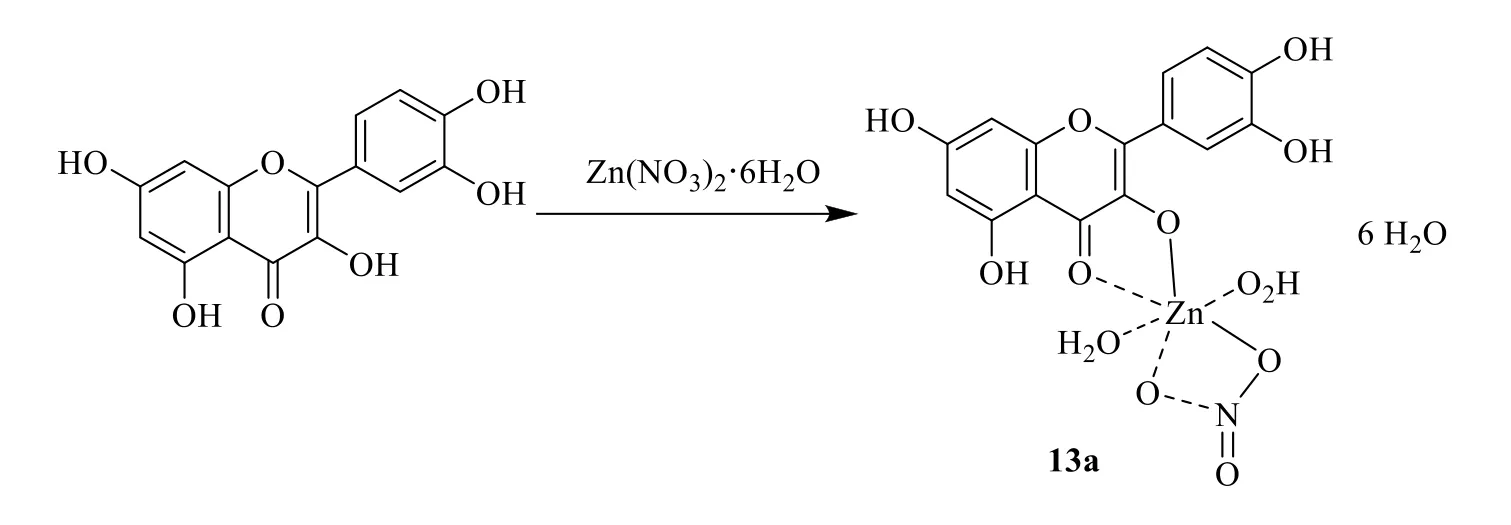
圖14 槲皮素鋅配合物的合成Fig.14 Synthesis of quercetin zinc complex
Lee 等[31]報道了一系列可激活過氧化物酶受體γ 的新型取代槲皮素衍生物。部分槲皮素衍生物的過氧化物酶體增殖物激活受體激動劑活性可與目前臨床使用的噻唑烷二酮類抗糖尿病藥物相媲美,結果表明取代槲皮素的噻唑烷二酮衍生物,如14i~14k 表現出良好的激動活性。化合物14k 具有不受保護的槲皮素單元,是該系列化合物中活性最強的,而甲氧甲基化的槲皮素衍生物14j 與丙二酸衍生物14m 和14n 不同,其活性較低。14m 和14k 將作為抗糖尿病藥物進一步研究。
槲皮素在DMF 中用過量的芐基溴,在K2CO3存在下得到14f。14f 在KH 存在下與14a 烷基化得到化合物14i。由于酚羥基需要不同的脫保護方法,因此選用甲氧甲基保護基團代替芐基保護酚羥基。以槲皮素和過量氯甲基甲醚為原料,合成了14g,并與14b 在KH 和NaI存在下偶聯,合成了14j。在微量鹽酸存在下,14j 在甲醇中回流,被裂解得到14k。14f 和1-溴-3-氯丙烷在KH 和NaI存在下反應得到14h。由14h 與14d 反應生成14l。14g 與14e反應生成14m。用微量的鹽酸在甲醇中除去14m 的甲氧甲基保護基,得到14n。用大于5 當量的2 mol/L氫氧化鋰水解14m,得到丙二酸衍生物14o。由14m在氫氧化銨存在下制備聯胺14p。14l 和14m 與碘甲烷甲基化分別得到α-甲基丙二酸酯類似物14q 和14r,見圖15。
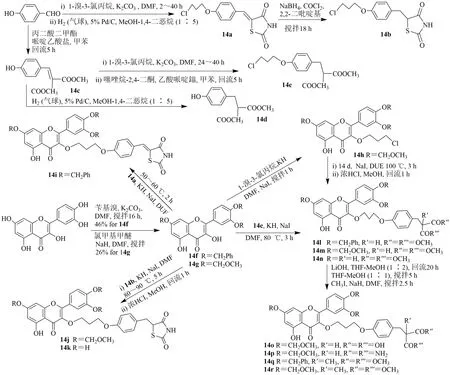
圖15 槲皮素-3-丙基醚衍生物的合成Fig.15 Synthesis of quercetin-3-propyl ether derivatives
1.3 槲皮素抗菌衍生物的合成
槲皮素與金屬離子配位后生物活性增加,溶解度和生物利用度增多[32]。銅是一種生物活性金屬,在生物過程中具有多種作用,例如催化大量生化反應并在線粒體中運輸電子起關鍵作用。銅(II)配合物顯示出多種生物活性,可以用作抗菌、抗炎、抗腫瘤和抗病毒作用,銅(II)配合物可作為潛在的治療藥物。槲皮素具有與多種金屬離子結合以增加其生物活性的能力。Moodi 等[33]使用槲皮素、乙醇胺和醋酸銅合成了槲皮素銅配合物,通過紅外光譜、氫譜和碳譜圖譜進行表征。光譜數據顯示槲皮素3-OH 和亞胺與金屬離子配位,結果表明該配合物對伯、仲醇的氧化表現出顯著的催化活性,對大腸桿菌(革蘭陰性菌)和金黃色葡萄球菌(革蘭陽性菌)有抗菌活性。由于細胞結構的差異金黃色葡萄球菌的抗菌活性高于大腸桿菌。
槲皮素溶于乙醇,冰醋酸、乙醇胺滴加到反應混合物中。在60 ℃下攪拌回流。將所得深紅色溶液濃縮冷卻,得到橙色結晶沉淀用熱乙醇溶液重結晶并干燥得到15a。將15a 溶解在去離子水中,加入NaOH 室溫攪拌,得到橙色溶液。滴加Cu(OAc)2去離子水溶液,超聲處理30 min。產生的棕色混合物后進行離心,并置于80 ℃的真空烘箱中6 h,得深橙色化合物15b,見圖16。
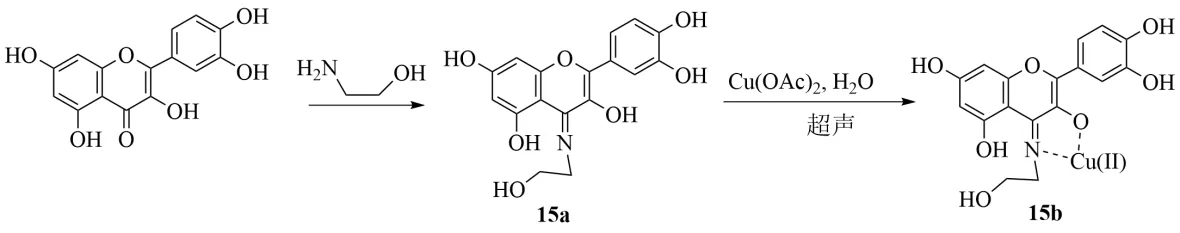
圖16 槲皮素銅配合物的合成Fig.16 Synthesis of quercetin copper complex
1.4 槲皮素抗炎衍生物的合成
Rasheed 等[34]通過研究槲皮素醋氯芬酸酯(16b)理化性質和藥理作用,旨在開發無潰瘍不良反應的新型非甾體抗炎藥。在模擬胃液、腸液和大鼠糞便中進行水解動力學研究,測定生化指標(胃壁黏液、己糖胺)、氧化參數(全血脂過氧化物、谷胱甘肽、過氧化氫酶、超氧化物歧化酶和蛋白質含量,結果表明,合成的16b 化學穩定,生物不溶性,具有較好的親脂性,同時保留了抗炎活性,潰瘍降低,胃腸道不良反應減少。與母藥相比,其蛋白質結合量小,吸收能力強,各項生化指標均表現出較好的效果。
醋氯芬酸(16x)加入氯化亞砜,室溫攪拌,減壓除去多余的氯化亞砜,得到黃色無定形固體醋氯芬酰氯(16a)。將蘆丁加入丙酮中,加入無水碳酸鉀和硫酸二甲酯,回流,濾過,減壓除溶劑,產物用乙醇硫酸回流,減壓脫除溶劑,乙醇重結晶得到16y。16y 溶解在含有三乙胺和4-二甲基氨基吡啶的二氯甲烷中,反應混合物冷卻至-10 ℃,將16a 溶于二氯甲烷,并將其在1 h 內逐滴加入,反應混合物攪拌過夜,減壓除去溶劑,乙醇重結晶獲得棕色16b,見圖17。
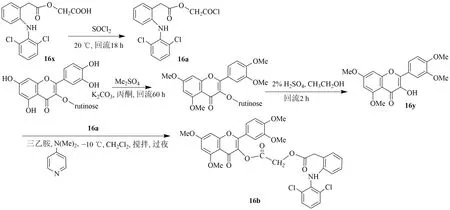
圖17 槲皮素醋氯芬酸酯的合成Fig.17 Synthesis of quercetin acetyl chlorphenate
Buravlev 等[35]通過曼尼希反應合成了4 個槲皮素-8-甲基氨基衍生物(17a~17d)。結果顯示,在抗壞血酸/Fe2+β 誘導的腦勻漿脂質過氧化模型中,所有衍生物均具有較高的抗氧化活性,17c 在低濃度下顯示出最高的活性。小鼠紅細胞氧化性溶血研究表明,具有嗎啉代甲基或硫代嗎啉代甲基的槲皮素衍生物具有保護原始細胞免受急性氧化應激和抗自由基活性,衍生物17c和17d在保護紅細胞免受H2O2誘導的急性氧化應激的能力上超過了槲皮素。因此,17c 具有較好的抗炎作用,可用于治療胃潰瘍。1,4-二氧六環的槲皮素溶液,冰浴,加入37%甲醛水溶液,然后加入胺。將混合物升溫至25 ℃,攪拌75 min(吡咯烷)或100 min(哌啶),濾過,用1,4-二氧六環洗滌干燥。在固體沉淀中加入2 mol/L HCl乙醇溶液,室溫下攪拌5 min。濾過,用2 mol/L 鹽酸乙醇溶液洗滌。真空中干燥,得黃色粉末產物17a或17b。將37%甲醛水溶液加入槲皮素的乙醇溶液中,加入嗎啉或硫代嗎啉,混合物在60 ℃加熱2 h,然后冷卻至室溫,濾過,用乙醇洗滌干燥。在固體沉淀中加入HCl 的乙醇溶液,攪拌后混合物在水浴中加熱回流,冷卻至室溫,濾過,用鹽酸的乙醇溶液洗滌,在真空中干燥,得到黃色粉末產物(17c 或17d),見圖18。
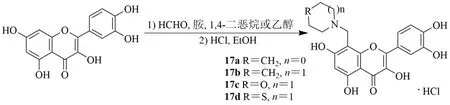
圖18 槲皮素-8-甲基氨基衍生物的合成Fig.18 Synthesis of quercetin-8-methylamino derivatives
1.5 槲皮素抗病毒衍生物的合成
為了尋找具有較高抗病毒活性的化合物,Han等[36]使用蘆丁和1,4-戊二烯-3-酮為原料合成了20個槲皮素衍生物,并通過半葉法評估其對煙草花葉病毒(tobacco mosaic virus,TMV)和黃瓜花葉病毒(cucumber mosaic virus,CMV)的抗病毒生物活性。結果表明20 種化合物對煙草花葉病毒顯示出良好的抗病毒活性。18r 對CMV 的治療活性高于其余化合物和寧南霉素。18k、18q~18t 表現出良好的滅活效果,優于蘆丁,低于寧南霉素。由于18q~18t 對CMV 顯示出良好的抑制活性,有望作為植物抗病毒劑得到進一步研究。實驗發現這些衍生物的抑制活性很大程度上取決于取代基的性質,當取代基為2-呋喃基或2-噻吩基時,相應的目標化合物對CMV 和TMV 表現出良好的抑制活性。
在蘆丁、K2CO3和丙酮溶液中,逐滴加入硫酸二甲酯,得到黃色膠狀固體18x。將18x 加入乙醇和鹽酸反應得到18y。18y 和K2CO3的混合物溶于DMF 中,加入氯醋酸乙酯反應得到黃色粉末18z;將18z 的甲醇溶液中滴加NaOH 溶液,得化合物18w;18w、DCC、DMAP 和含不同的取代基團的1,4-戊二烯-3-酮18A~18T 溶解在二氯甲烷中反應,過色譜柱,醋酸乙酯和石油醚洗脫,乙醇重結晶,得到目標化合物18a~18t,見圖19。
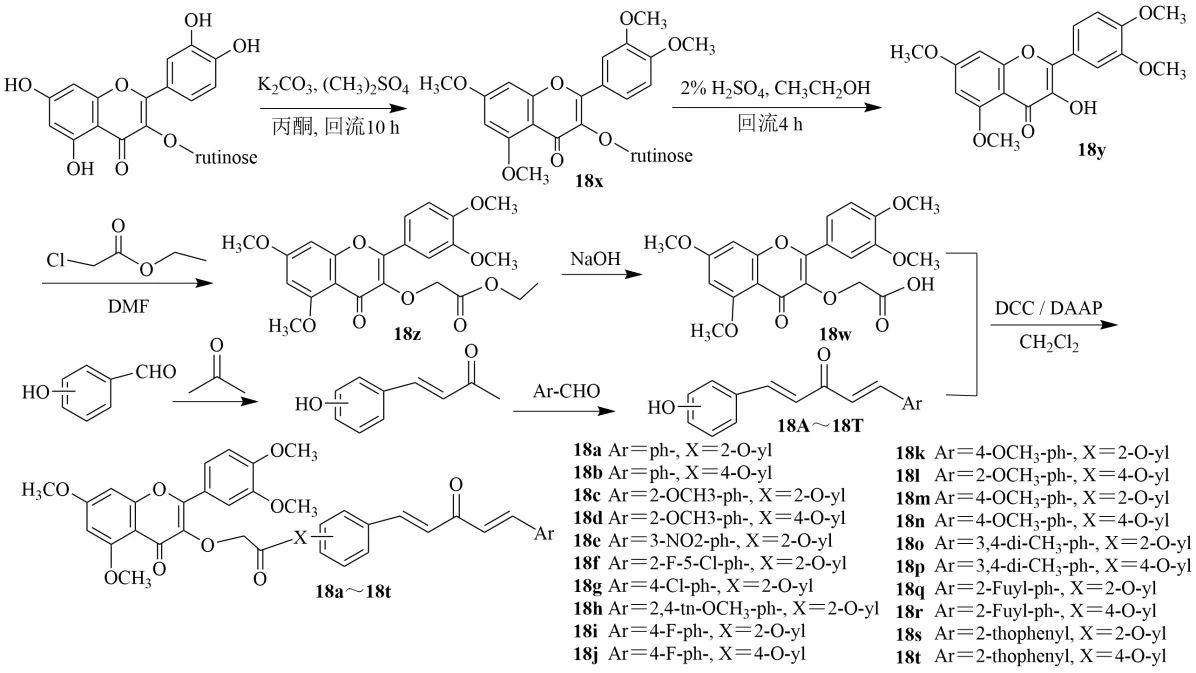
圖19 含有1,4-戊二烯-3-酮槲皮素衍生物的合成Fig.19 Synthesis of quercetin derivatives containing 1,4-pentadien-3-one
丙型肝炎病毒二酮酸(α,γ-diketoacid,DKA)的類似物或異構體通過螯合活性位點上的2 個鎂離子,對丙型肝炎病毒NS5B 聚合酶有很強的抑制作用。槲皮素的抗丙型肝炎病毒(hepatitis C virus,HCV)活性部分歸因于其是DKA 的結構模擬物。為了揭示槲皮素抑制HCV 活性所需的結構特征,提高抗HCV 活性,Zhong 等[37]、Lee 等[38]設計、合成了19 種槲皮素-7-O-芐基衍生物(19a~19s),并在細胞檢測中對其抗HCV 性能進行評價,結果表明合成的槲皮素衍生物均表現出強效抗HCV 活性,其EC50值范圍為3.8~8.7 μmol/L。在這些化合物中,間氯取代衍生物具有最高的活性(19i,EC50=3.8 μmol/L)。槲皮素在7-O 位的取代基可能類似于DKA 的間位取代基,從而增強抗HCV 活性。分子對接研究表明,槲皮素衍生物能夠與2 個鎂離子建立關鍵配位,并與丙型肝炎病毒NS5B 活性位點的殘基相互作用。
槲皮素與醋酸酐反應得19x,19x 在NMP 中與硫酚和咪唑進行區域選擇性脫乙酰化,得到19y。19y 與不同取代的芐基溴化物烷基化,然后用甲醇氨處理脫乙酰基,得到19a~19s,見圖20。
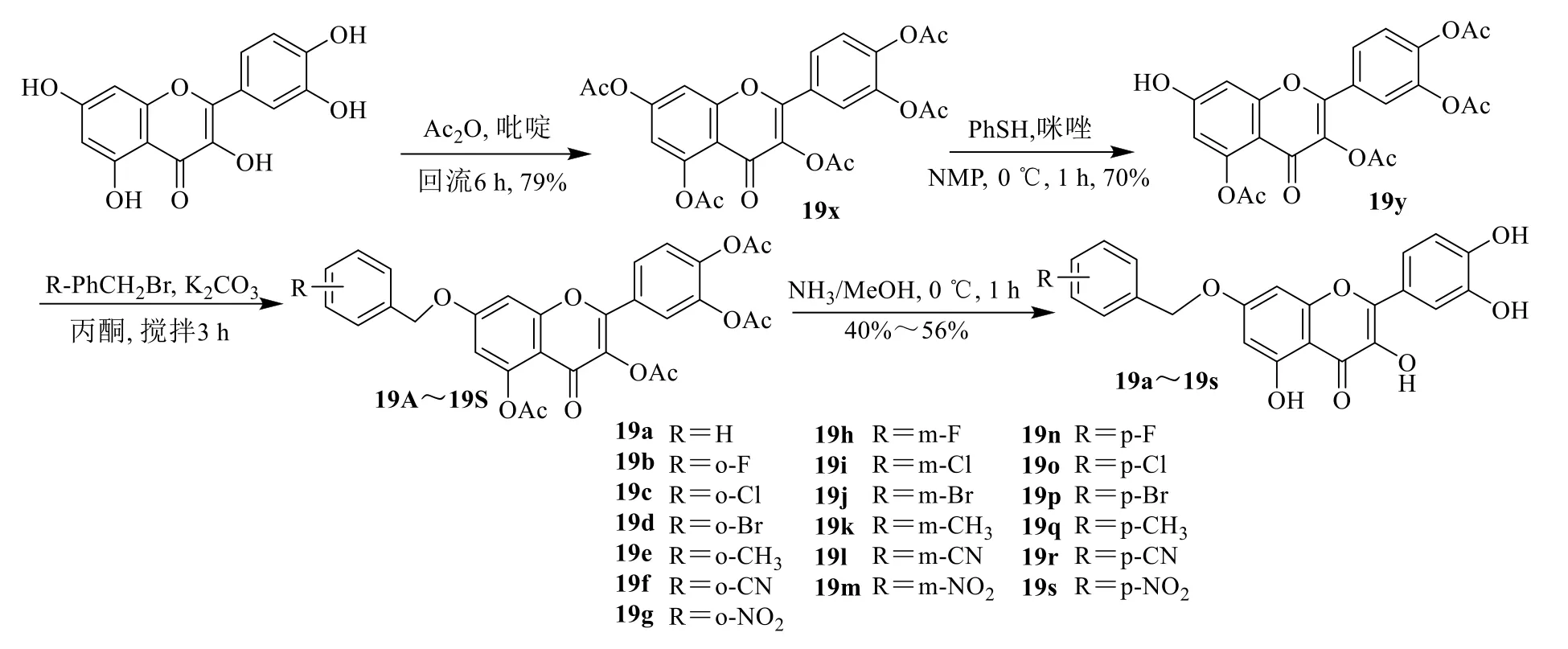
圖20 槲皮素-7-O-芐基衍生物的合成Fig.20 Synthesis of quercetin-7-O-benzyl derivatives
2 構效關系
具有核心類黃酮骨架的天然產物已被證明是生物活性化合物的良好來源。大量研究表明,含有黃酮類化合物的天然產物可以克服多種MDR 細胞[39-40]。槲皮素具有C6-C3-C6碳骨架結構,由2 個苯環A 和B 組成,并通過三碳吡喃環C 連接。在槲皮素分子中,B 環存在鄰二酚結構,A 環有間二酚結構,C 環有1 個烯醇式、羥基酮結構,這些結構使得槲皮素具有一些特殊的生物活性。槲皮素的3 個環是平面的,在該體系中,分子間形成3 個氫鍵:2 個氫鍵通過羰基團建立,另1 個在B 環的OH 基團之間形成。槲皮素因其顯著的抗氧化和抗癌活性而被廣泛研究,修飾不同基團得到的衍生物具有不同的生物活性和功效[41-42],見圖21。
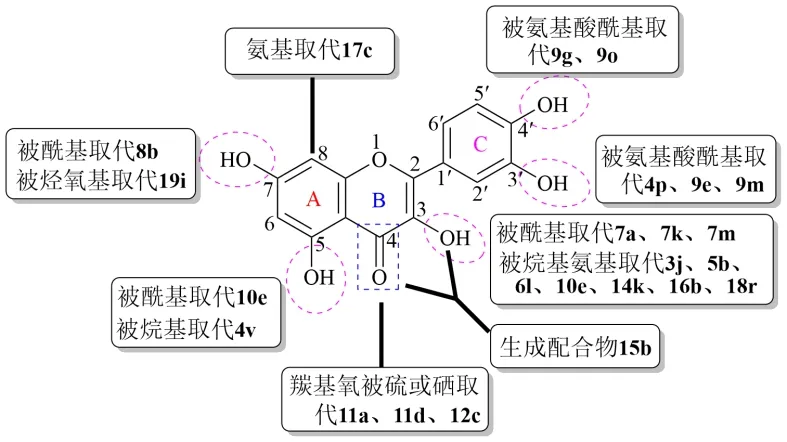
圖21 活性較好的槲皮素衍生物Fig.21 More active quercetin derivatives
通過比較槲皮素及其衍生物的活性,可以發現
衍生物的構效關系主要取決于取代基的位置和性質。槲皮素的3?-OH 是非常重要的位置,引入N-甲基-N-丙基哌嗪(4p),油水分配系數增大,水溶性提高,細胞穿透能力增強,細胞毒性活性增加了96倍,IC50值為0.48 μmol/L,在結腸癌模型中,CT-26荷瘤小鼠體內治療17 例,與槲皮素相比,顯著提高了存活率并減輕了腫瘤重量(60%)[18]。槲皮素-3?-O-甘氨酸氨基甲酸酯是一種水溶性前藥,仍需進一步的臨床研究,以適合癌癥患者使用[43]。對槲皮素3?-OH 進行修飾,抗癌活性顯著增強[24]。對槲皮素的3-OH 或者5-OH 進行修飾,可提高其抗病毒、抗糖尿病、抗癌和抗炎能力[20-22,26,31,36]。在槲皮素中引入羧基或酯基,可提高脂溶性、生物利用度和抗腫瘤活性[22-23];槲皮素的羰基氧被硫或硒取代,抗腫瘤活性增強[27-28];槲皮素金屬配合物有較好的抗菌、抗糖尿病活性[30,33]。通過曼尼希反應,在槲皮素8位引入不同的基團,可以改善口服利用度,可開發為治療胃潰瘍的藥物[35]。
3 結語
槲皮素是一種有價值的天然黃酮類化合物,由于能夠調節多種靶點和信號通路而得到了廣泛研究。由于槲皮素的低溶解性和生物利用度限制了其應用[44-45]。因此,設計和合成新的槲皮素衍生物來改變其局限性勢在必行。目前,對槲皮素的結構進行優化修飾,已經合成了許多溶解性能好、生物利用度高的槲皮素衍生物,在抗癌、抗氧化/抗衰老、抗病毒、抗炎、降糖等方面有許多優勢,應用前景廣闊。希望更多的研究者積極參與其中,使天然藥物經過優化修飾后早日進入臨床,發揮天然藥物的作用,為患者解除痛苦。
利益沖突所有作者均聲明不存在利益沖突
