氣相色譜-質譜聯用技術測定水中低濃度揮發性有機化合物濃縮富集水平研究
2023-05-30 22:58:22徐小培吳飛周樂吳晨熊文夏俊鵬
食品安全導刊 2023年5期
關鍵詞:氣相色譜
徐小培 吳飛 周樂 吳晨 熊文 夏俊鵬

摘 要:由塑化劑產生的揮發性有機物(Volatile Organic Compounds,VOCs)引起的水體污染問題是當前研究的熱點。由于飲用水中VOCs濃度較低,因此需選擇科學的樣品前處理手段及靈敏度較高的檢測手段。本研究采用氣相色譜-質譜法對水體中的低濃度VOCs進行富集,通過對實驗條件進行優化,實現對水體中VOCs的高靈敏快速分析。實驗結果表明59種微量揮發性有機化合物的線性相關系數均大于0.999,方法檢出限為0.02~0.09 μg·L-1,加標回收率為85.9%~107.0%,相對標準偏差為0.3%~6.2%,使用該方法測定飲用水中低濃度揮發性有機物,結果準確且方便快捷。
關鍵詞:氣相色譜-質譜聯用技術;揮發性有機化合物;濃縮富集水平;吹掃捕集
Abstract: The problem of water pollution caused by volatile organic compounds (VOCs) produced by plasticizers is a hot topic of current research. Due to the low concentration of VOCs in drinking water, it is necessary to choose scientific sample pretreatment methods and sensitive detection methods. This study used gas chromatography-mass spectrometry to enrich low concentration VOCs in water, and optimized experimental conditions to achieve highly sensitive and rapid analysis of VOCs in water. The experimental results show that the linear correlation coefficients of 59 trace volatile organic compounds are all greater than 0.999, and the detection limit of the method is 0.02~0.09 μg·L-1, with a recovery rate of 85.9%~107.0% and a relative standard deviation of 0.3%~6.2%. The method is accurate, convenient, and fast for the determination of low concentration volatile organic compounds in drinking water.
Keywords: gas chromatography mass spectrometry technology; volatile organic compounds; concentration and enrichment level; purging and trapping
近年來,揮發性有機物(Volatile Organic Compounds,VOCs)引起的水體環境污染日益引起人們的重視。企業污染物的排放和使用化學產品等均可導致VOCs的產生,目前已知的VOCs有700多萬種,其中,人工合成的VOCs有10萬種以上,且以每年2 000多種的速度增長[1]。VOCs在生產、銷售、儲存、加工和使用等環節極易進入環境中,特別是水環境中,進而引發水污染。大多數VOCs有強烈的致癌性、致畸性和致突變性等毒性機制[2-3]。因此,對水中 VOCs進行準確檢測顯得尤為重要。由于水體中VOCs含量很少,且成分復雜,沸點較低,因此在進行VOCs檢測前需要先對其進行富集。VOCs富集技術按其處理方式可劃分為液液萃取、靜態頂空萃取、吹掃捕集、固相微萃取、膜萃取、蒸餾和直接注入等[4-5]。其中吹掃捕集應用最為廣泛,與氣相色譜-質譜法聯用可實現對水體中多種VOCs的精確定性、定量分析[6]。因此,本研究采用凈化捕集技術對水中揮發性有機物進行預處理,通過優化水中59種揮發性有機物的吹掃捕集條件、氣相色譜條件和質譜條件,建立了吹掃捕集-氣相色譜-質譜聯合測定水中揮發性有機物的高效、靈敏的方法。
1 材料與方法
1.1 儀器與試劑
6890N型氣相色譜儀,安捷倫公司;5973i型質譜儀,安捷倫公司;吹掃捕集裝置(Lumin型),Tekmar公司。54種揮發性有機物混合標準溶液(質量濃度均為200 mg·L-1,上海安譜實驗科技股份有限公司);氯甲烷、氯乙烯、溴甲烷、氯乙烷混合標準溶液(質量濃度均為2 000 mg·L-1,美國化學服務公司);甲基叔丁基醚標準溶液(2 000 mg·L-1,上海安譜實驗科技股份有限公司);4-溴氟苯、1,2-二氯苯-d4混合標準溶液(質量濃度均為2 000 mg·L-1,上海安譜實驗科技股份有限公司);甲醇(色譜純);鹽酸(優級純);二次蒸餾水。
1.2 實驗方法
1.2.1 標準溶液配制
(1)59種VOCs混合標準中間液1(質量濃度均為20.0 mg·L-1)。將100 μL 54種VOCs混合標準溶液和10 μL氯甲烷、氯乙烯、溴甲烷、氯乙烷混合標準溶液,以及10 μL甲基叔丁基醚標準溶液置于1 mL棕色容量瓶中,用甲醇定容至刻度,搖勻。
(2)59種VOCs混合標準中間液2(質量濃度均為2.0 mg·L-1)。將100 μL 59種VOCs混合標準中間液1置于1 mL棕色容量瓶中,用甲醇定容至刻度,混勻。
(3)4-溴氟苯、1,2-二氯苯-d4混合標準中間液(質量濃度均為5.0 mg·L-1)。移取4-溴氟苯、1,2-二氯苯-d4混合標準溶液25 μL于10 mL棕色容量瓶中,用甲醇定容至刻度,混勻。
(4)59種VOCs系列混合標準工作溶液。分別移取12.5 μL、25.0 μL 59種VOCs混合標準中間液2和12.5 μL、25.0 μL、62.5 μL、125.0 μL 59種VOCs混合標準中間液1,置于6只50 mL棕色容量瓶中,各加入4-溴氟苯、1,2-二氯苯-d4混合標準中間液100 μL,用水定容至標線,混勻,配制成系列混合標準工作溶液,其中59種揮發性有機物的質量濃度均分別為0.50 μg·L-1、1.00 μg·L-1、5.00 μg·L-1、10.00 μg·L-1、25.00 μg·L-1和50.00 μg·L-1,替代物4-溴氟苯、1,2-二氯苯-d4的質量濃度均為10.0 μg·L-1。系列混合標準工作溶液配制完成后裝入40 mL帶聚四氟乙烯墊片的進樣瓶中,裝滿,上方不留空氣,上機等待進樣。
1.2.2 儀器條件
(1)氣相色譜條件。DB-5MS柱(60 m×0.25 mm,0.25 μm,美國安捷倫科技有限公司);進樣口溫度:200 ℃;進樣方式:分流進樣,分流比為1∶20;進樣體積:1.0 mL;載氣:高純氦氣,純度不小于99.999%(體積分數);柱流量:1.0 mL·min-1;升溫程序:初始溫度為40 ℃,保持5.0 min,以6 ℃·min-1升溫至200 ℃,保持2.0 min。
(2)吹掃捕集條件。吹掃溫度:20 ℃;吹掃流量:40 mL·min-1;吹掃時間:11.0 min;干吹掃時間:0.5 min;預解析溫度:245 ℃;解析溫度:250 ℃;解析時間:2.0 min;烘烤溫度:260 ℃;烘烤時間:2.0 min。
(3)質譜條件。電子轟擊源:EI;離子化能量:70 eV;離子源溫度:230 ℃;傳輸線溫度:270 ℃;質量掃描范圍:45~350 Da;溶劑延遲時間:4.4 min;掃描方式:選擇離子掃描(SIM)模式。
2 結果與分析
2.1 色譜條件優化
色譜柱溫度是一個重要指標,對樣品中待測物質的分離效果及分析速率有較大的影響。在程序升溫過程中,初始溫度一般比室溫略高,經多次試驗發現,選擇初始溫度為40 ℃并保持5 min,即可使較早地出現色譜峰,并且色譜峰具有良好的分離度。若溫度過低,則會有殘留物質滯留于色譜柱上,且不利于儀器達到溫度平衡。升溫速率對中間出峰物質的分離度有較大影響,柱升溫速率較小時,分析時間相對較長,提高柱溫可以促使氣相傳質速率加快,但會導致柱效下降,因此在兼顧難分離組分的分離度及色譜峰形不拖尾的前提下選擇了較低的柱溫。相關文獻顯示[7],為了實現對一些復雜物質的分離,VOCs一般會設置多個升溫速率,但是在適用的物質類型上存在局限性。在此基礎上,針對VOCs種類繁多和普適性的特點,選擇1.2.2氣相色譜條件能夠滿足59種VOCs和2種替代物的定性分析要求。在這種色譜條件下,只需要一次程序升溫,就可以對59種VOCs進行有效的分離和定性,而且簡單、高效、易于使用。
2.2 吹掃捕集條件優化
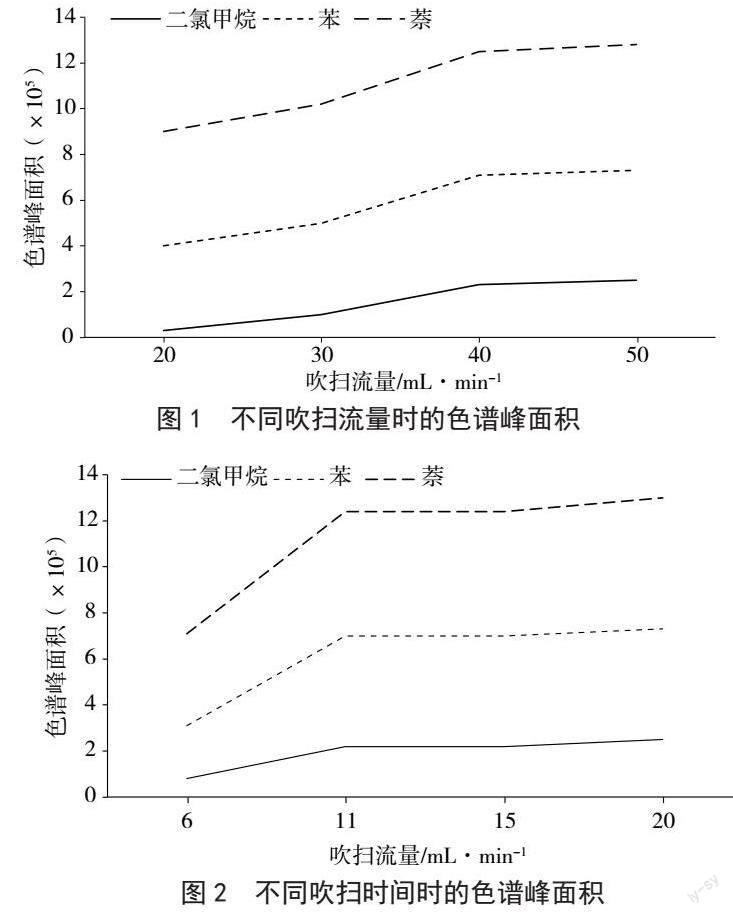
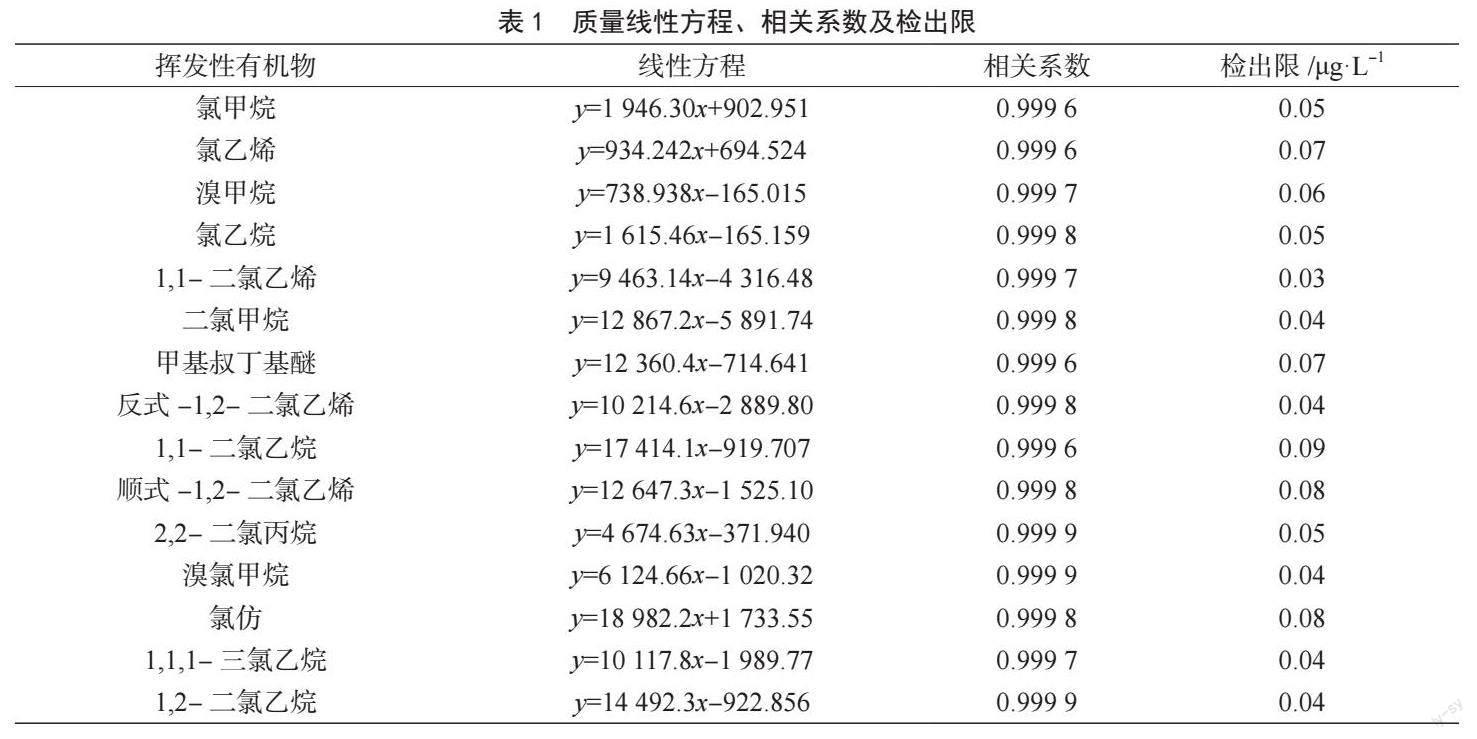
吹掃捕集條件的優化以二氯甲烷、苯和萘為例,固定吹掃時間為11 min,分別觀察三者吹掃流量為20 mL·min-1、30 mL·min-1、40 mL·min-1和50 mL·min-1時的色譜峰面積。圖1結果表明,在吹掃流量達到40 mL·min-1后,色譜峰面積均保持在相對穩定的狀態,繼續增大吹掃流量對色譜峰面積無明顯的影響,因此選擇吹掃流量為40 mL·min-1。吹掃流量為40 mL·min-1時,比較吹掃時間對色譜峰面積的影響。圖2結果表明,在吹掃時間達到11 min后,色譜峰面積逐漸穩定,因此選擇吹掃時間為11 min。
2.3 替代物選擇
替代物是樣品中不含有但其物理化學性質與待測目標化合物相似的物質。一般在樣品提取或其他預處理之前加入,用來監控試驗過程中目標物是否有損失,以其回收率可以判斷或評價樣品基體、樣品處理過程對分析結果的影響,同時也能反映儀器的狀態變化。本研究選擇4-溴氟苯、1,2-二氯苯-d4為替代物。
2.4 線性關系與檢出限
在1.2.2儀器工作條件下,對系列混合標準工作溶液進行測定,將質量濃度設為橫坐標(x)、對應的色譜峰面積設為縱坐標(y)進行線性回歸,計算線性方程和相關系數。按樣品分析全流程步驟,以空白水樣代替實際樣品,平行制備7份加標質量濃度均為0.50 μg·L-1的樣品并測定,計算檢出限。由表1可知,59種VOCs在0.50~50.00 μg·L-1線性相關系數均大于0.999,線性關系良好,檢出限為0.02~0.09 μg·L-1。
2.5 加標回收與精密度實驗
取5 mL水樣,分別加入適量的59種VOCs混合標準中間液1和59種VOCs混合標準中間液2及4-溴氟苯、1,2-二氯苯-d4混合標準中間液,配制成低、中、高3種濃度水平的加標樣品,其中目標物質量濃度分別為1.00 μg·L-1、10.00 μg·L-1、40.00 μg·L-1,替代物質量濃度為10.0 μg·L-1。不同濃度的樣品均制備7份,進行測定。結果顯示樣品的加標回收率為85.9%~107.0%,相對標準偏差為0.3%~6.2%。表明該方法精密度、準確度均滿足分析要求。
3 結論
本研究利用吹掃捕集-氣相色譜-質譜聯用法對水中59種低濃度VOCs進行測定,結果顯示,59種VOCs的線性相關系數均大于0.999,方法檢出限為0.02~0.09 μg·L-1,加標回收率為85.9%~107.0%,相對標準偏差為0.3%~6.2%,提示該測定方法可以準確測定水中低濃度VOCs,具有檢出限低、精密度高且操作簡單的優勢,能夠滿足大批量水質樣品中多種VOCs的測定。
參考文獻
[1]陳君君,徐鳳.生活飲用水中揮發性有機物檢測方法的研究[J].中國城鄉企業衛生,2019,34(4):81-83.
[2]郭玉華.環境檢測中揮發性有機物檢測方法[J].黑龍江環境通報,2021,34(3):20-21.
[3]史芳.飲用水中揮發性有機物的檢測分析[J].化工設計通訊,2021,47(1):68-69.
[4]李庶峰.利用頂空氣相色譜方法檢測環境中的揮發性有機物[J].化學工程師,2022,316(1):26-29.
[5]李秋萱,劉玲花,王學東,等.地下水中揮發性有機物的樣品預處理與分析檢測綜述[J].首都師范大學學報(自然科學版),2022,43(1):91-96.
[6]林偉強,李美惠.吹掃捕集–氣相色譜/質譜聯用法測定飲用水中28種揮發性有機化合物[J].供水技術,2019,13(6):49-52.
[7]付瑤,張利鈞,楊豐春,等.吹掃捕集–氣相色譜–質譜法快速測定同序列地下水和土壤中的37中揮發性有機物[J].理化檢驗(化學分冊),2021,57(8):673-681.
猜你喜歡
中國纖檢(2016年12期)2017-01-20 09:28:19
現代農業科技(2016年20期)2016-12-20 14:51:09
現代農業科技(2016年20期)2016-12-20 09:05:36
分析化學(2016年7期)2016-12-08 00:09:44
分析化學(2016年7期)2016-12-08 00:07:08
價值工程(2016年29期)2016-11-14 01:34:54
科技視界(2016年24期)2016-10-11 18:58:00
考試周刊(2016年39期)2016-06-12 16:01:44
中國科技博覽(2016年4期)2016-04-25 07:25:47
中國科技博覽(2016年8期)2016-04-25 04:57:50
