HPLC-DAD法測定獸藥恩諾沙星注射液中非法添加呋塞米
2023-10-26 12:21:38陳錫龍楊秀玉孫真崢王慶紅謝麗麗張秀英
中國獸藥雜志 2023年9期
陳錫龍,楊秀玉,趙 貴,孫真崢,王慶紅,謝麗麗,楊 強,張秀英*
(1.貴州省獸藥飼料檢測所,貴陽 550003;2.中國獸醫藥品監察所,北京 100081)
恩諾沙星注射液和呋塞米注射液均收載于《中國獸藥典》2020年版一部[1]。恩諾沙星注射液為喹諾酮類獸醫專用抗菌藥,主要用于畜禽細菌性疾病和支原體感染,臨床應用廣泛;呋塞米注射液為利尿藥,臨床主要用于治療各種水腫癥。實踐中聯合使用恩諾沙星和呋塞米治療仔豬水腫病[2-3],據稱效果明顯。恩諾沙星和呋塞米復方注射液曾申請國家發明專利[4-5],但未獲授權,同時,該制劑不符合國家獸藥管理相關政策,也未獲生產許可,故在恩諾沙星注射液及其相應制劑中添加呋塞米或在呋塞米注射液及其相應制劑中添加恩諾沙星均屬非法添加。2022年第二季度,貴州省獸藥飼料檢測所發現1批獸藥恩諾沙星注射液中疑似添加呋塞米,違反《獸藥管理條例》,存在較大安全隱患。功能食品[6]、保健食品[7]、中成藥[8-10]、酵素[11]、減肥藥[12]和減肥食品[13-17]等均有報道可能存在非法添加呋塞米的情況,并建立了相應的檢測方法。非法添加呋塞米的檢測方法主要為超高效液相色譜-串聯質譜檢測法,此外,還有電噴霧-離子遷移譜法[15]、薄層色譜-近紅外光譜(TLC-NIRS)檢測方法[18]、酶聯免疫法[19]等。在此之前,農業農村部尚未發布獸藥中非法添加呋塞米的檢查方法。據此,貴州省獸藥飼料檢測所首先對其相關檢測方法進行了探索研究,并函請中國獸醫藥品監察所對方法和結果進行確認。根據農業農村部公告第289號“附件1:獸藥中非特定添加物質檢查方法”,同時參考《中國獸藥典》2020年版一部 “呋塞米注射液”項下的含量測定方法,選擇呋塞米為測試藥物,恩諾沙星注射液為目標制劑,建立了恩諾沙星注射液中非法添加呋塞米的檢查方法(液相色譜-二極管陣列法),為嚴厲打擊非法添加呋塞米提供技術支撐。該方法已經農業農村部公告第611號發布。
1 材料與方法
1.1 材料與試劑
1.1.1 試劑 乙腈為色譜純,水為超純水,四氫呋喃、冰醋酸為分析純。
1.1.2 試藥
1.1.2.1 測試藥物 呋塞米對照品,中國食品藥品檢定研究院,批號:100544-202104,含量:99.8%。
1.1.2.2 制劑空白樣品 恩諾沙星注射液;生產單位:安徽天安生物科技有限公司;批號為20210625;規格:10 mL:0.25 g。
1.1.2.3 供試品 恩諾沙星注射液,生產單位:X公司;批號:20210501,規格:10 mL:0.25 g。經檢測含有呋塞米。
1.1.2.4 陽性模擬樣品 制劑空白樣品(1.1.2.2)中添加測試藥物(1.1.2.1)呋塞米,作為陽性模擬樣品。
1.2 儀器與設備 Thermo Scientific U3000高效液相色譜儀(配DAD檢測器);分析天平(XSR205DU型,分度值0.01 mg, Mettler)。
1.3 實驗方法
1.3.1 色譜條件 色譜柱:Hypersil GOLD C18,4.6×250 mm 5 μm;流動相:水-四氫呋喃-冰醋酸(70∶30∶1);檢測波長:二極管陣列檢測器采集波長范圍為200~400 nm;分辨率為1.0 nm;記錄272 nm波長處的色譜圖;柱溫:30 ℃;流速:1.0 ml/min;進樣量:20 μL。
1.3.2 試驗溶液
1.3.2.1 溶媒 參考《中國獸藥典》2020年版一部中呋塞米注射液含量測定方法用的混合溶液,即冰醋酸-乙腈-水(22∶489∶489),作為呋塞米的溶媒。
1.3.2.2 各種溶液的制備
1.3.2.2.1 空白溶液 ①制劑空白溶液:取恩諾沙星注射液空白制劑1.00 mL,置20 mL量瓶中,加混合溶劑[取冰醋酸22 mL,加乙腈-水(1∶1)至1000 mL,混勻]稀釋定容;另取2.00 mL,置20 mL量瓶中,加混合溶劑稀釋至刻度,搖勻,即得;② 溶劑空白:混合溶液。
1.3.2.2.2 對照品溶液 ① 呋塞米對照品貯備液:取呋塞米對照品約25 mg,精密稱定,置50 mL量瓶中,加混合溶液使溶解并稀釋至刻度,搖勻(0.5 mg/mL);② 呋塞米對照品溶液:取呋塞米對照品貯備液2.00 mL,置20 mL量瓶中,用混合溶液使溶解并稀釋至刻度,搖勻(0.05 mg/mL);③ 標準曲線系列溶液的配制:精密量取呋塞米對照品貯備液適量,加混合溶劑分別制成2.0、5.0、10.0、25.0、50.0和100.0 μg/mL的標準曲線系列溶液。
1.3.2.2.3 供試品溶液 取供試品1.00 mL,置20 mL量瓶中,加混合溶劑[取冰醋酸22 mL,加乙腈-水(1∶1)至1000 mL,混勻]稀釋定容;另取2.00 mL,置20 mL量瓶中,加混合溶劑稀釋至刻度,搖勻,作為供試品溶液。
1.3.2.2.4 陽性模擬樣品溶液 陽性添加樣品溶液的配制:取制劑空白樣品1.00 mL,置20 mL量瓶中,按高(H)、中(M)、低(L)三個濃度水平分別加入10、8和6 mL濃度為0.5 mg/mL的呋塞米對照品貯備液,混勻,加混合溶劑稀釋至刻度,搖勻;另取2.00 mL,置20 mL量瓶中,加混合溶劑稀釋至刻度,搖勻,作為陽性添加樣品溶液。
1.3.2.2.5 檢測限溶液的配制 取制劑空白樣品1.00 mL,置20 mL量瓶中,分別加入0.5 mg/mL的呋塞米對照品貯備液0.2、0.4 和0.8 mL,混勻,加混合溶劑稀釋至刻度,搖勻;分別另取2.00 mL,置20 mL量瓶中,加混合溶劑稀釋至刻度,搖勻,分別制成0.5、1.0、2.0 μg/mL的檢測限溶液。
1.3.3 測定法 精密量取各試驗溶液20 μL注入高效液相色譜儀測定,同時記錄色譜圖與光譜圖。
2 結果與分析
2.1 色譜條件
2.1.1 流動相 采用《中國獸藥典》2020年版一部中呋塞米注射液的含量測定的高效液相色譜法的流動相“水-四氫呋喃-冰醋酸(70∶30∶1)”系統作為試驗的流動相。
2.1.2 提取波長 照1.3.1項下色譜條件,進樣呋塞米對照品溶液,得到的光譜圖中有三個最大吸收波長,分別為236、274和344 nm,考慮到《中國獸藥典》2020年版一部中呋塞米注射液的含量測定波長為272 nm,故以272 nm為提取波長。
2.2 方法學考察
2.2.1 專屬性試驗 干擾考察。分別取溶劑空白、制劑空白溶液、供試品溶液、陽性模擬樣品溶液和對照品溶液各20 μL,照1.3.1項下色譜條件測定,恩諾沙星注射液本底及溶劑對被測物呋塞米的測定沒有干擾。按擬定檢查方法進行檢測,結果表明在呋塞米出峰處無干擾峰,且達到基線分離,專屬性良好(圖1)。
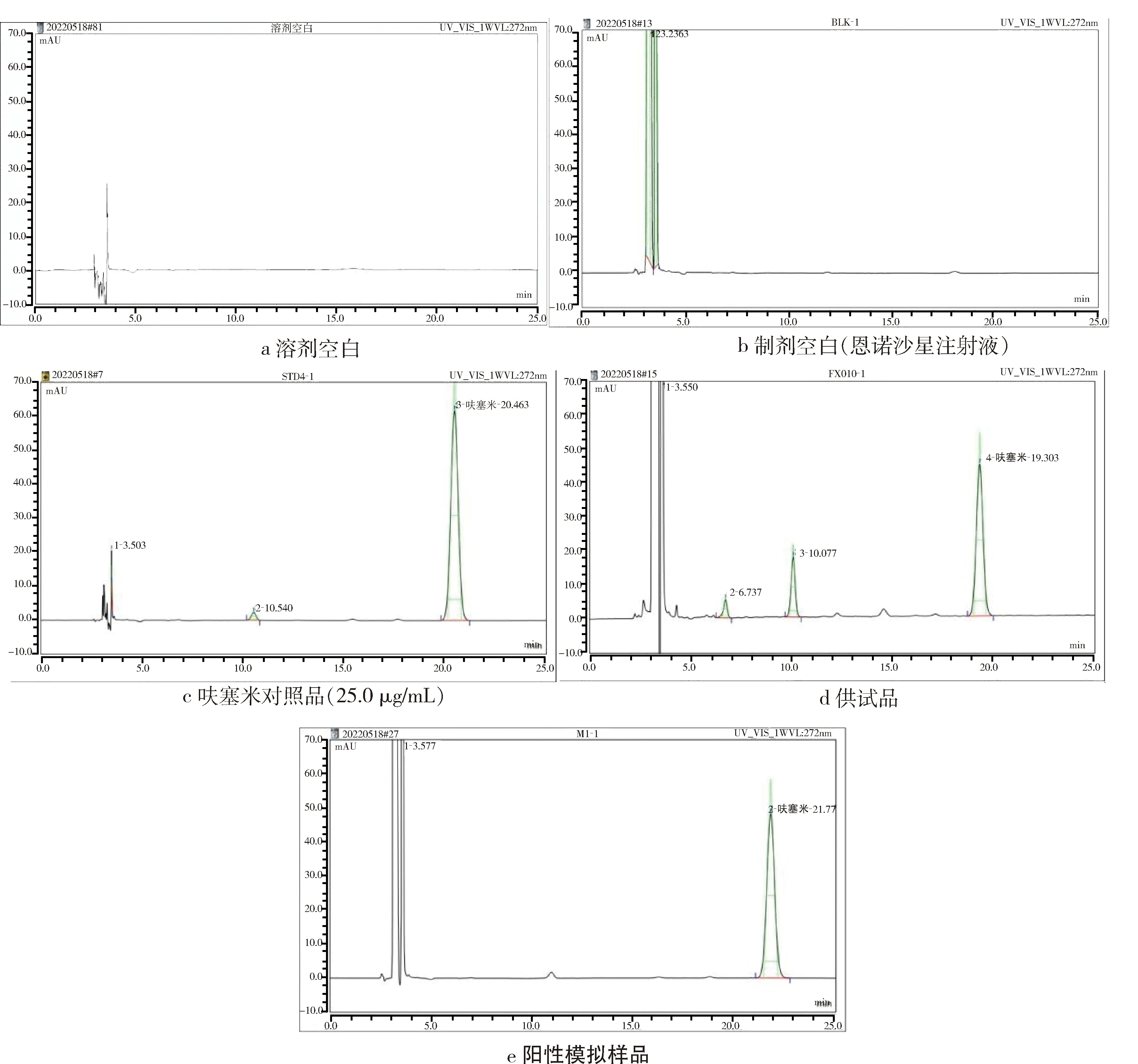
圖1 溶劑空白、制劑空白、呋塞米對照品、供試品及陽性模擬樣品色譜圖Fig 1 Chromatograms of solvent、Enrofloxacin Injection、reference furosemide、sample and positive simulation sample
2.2.2 峰純度和光譜相似度檢查 通過供試品和呋塞米對照品溶液試驗進行峰純度和光譜相似度檢查。分別精密量取1.3.2.2.3項下的供試品溶液和1.3.2.2.2項下的呋塞米對照品溶液各20 μL,按1.3.1項下的色譜條件,注入高效液相色譜儀,采集3D數據,記錄光譜圖(圖2、圖3)。結果表明,峰純度分析顯示,供試品和對照品峰匹配(表示峰最大值處的光譜與前沿和拖尾上光譜的相似度)值均為1000(匹配值大于980可以基本確認是同一組分),說明呋塞米峰為單一組分。經目測比較,可知供試品和對照品光譜圖一致(最大吸收波長偏差不超過2 nm)。或者,通過執行光譜跟蹤把供試品和對照品的光譜圖與用呋塞米對照品溶液建立的光譜庫進行匹配,相似度閾值設為980,可知供試品和對照品與呋塞米光譜庫的相似度分別為1000和998,即它們的紫外光譜一致。
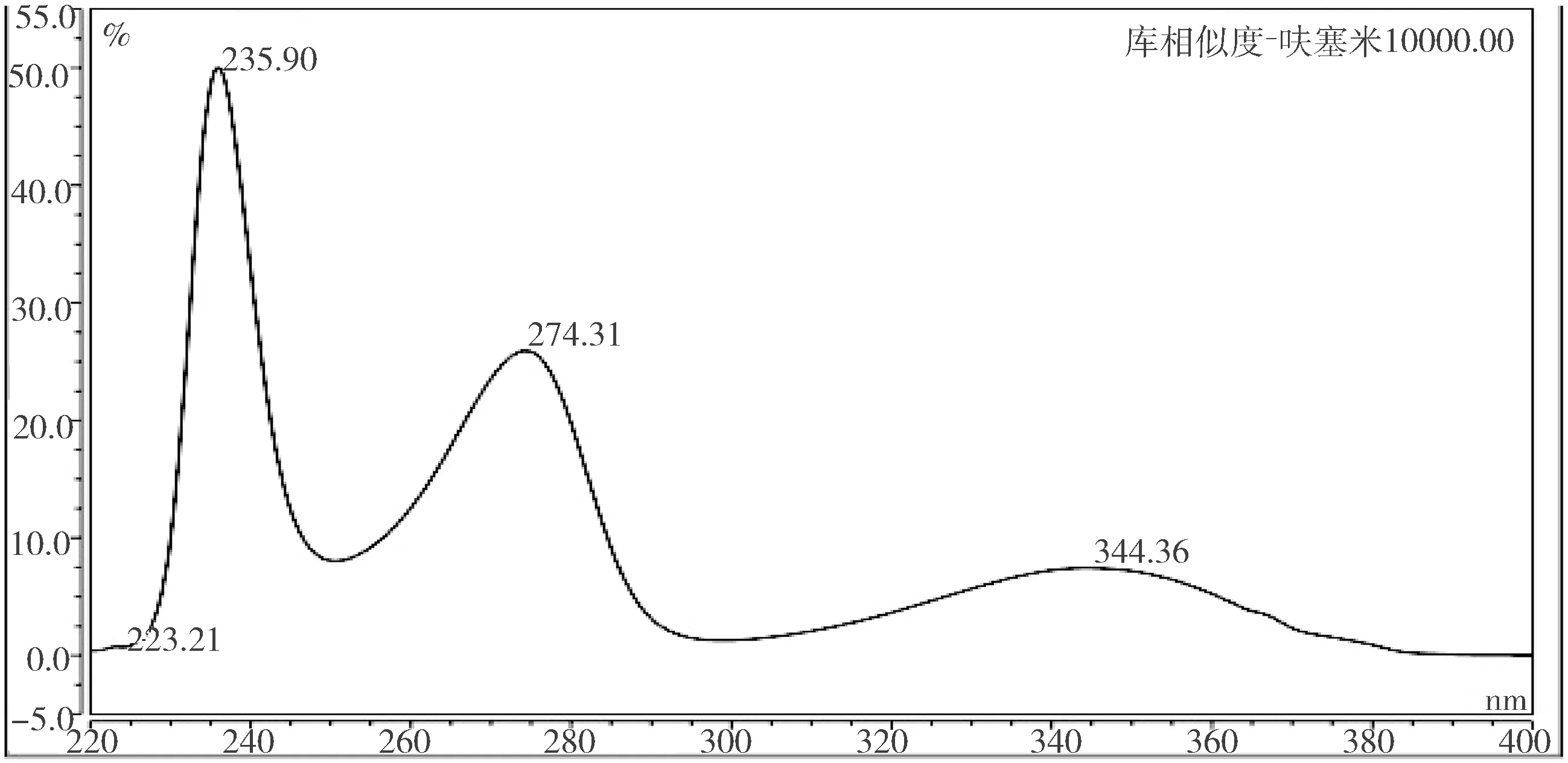
圖2 呋塞米對照品光譜圖Fig 2 The spectrum of reference furosemide
圖3 供試品光譜圖Fig 3 The specrum of the sample
2.2.3 線性與范圍 線性關系考察:吸取1.3.2.2.2項③中的標準曲線系列溶液20 μL進行測定,每個濃度平行進樣兩次。以呋塞米峰面積Y為縱坐標,相應濃度X為橫坐標,繪制標準曲線。回歸方程為:Y=1.0629X+0.0685,R2=1.0000。結果表明,呋塞米在2.0~100.0 μg/ml范圍內線性關系良好。結果見表1和圖4。
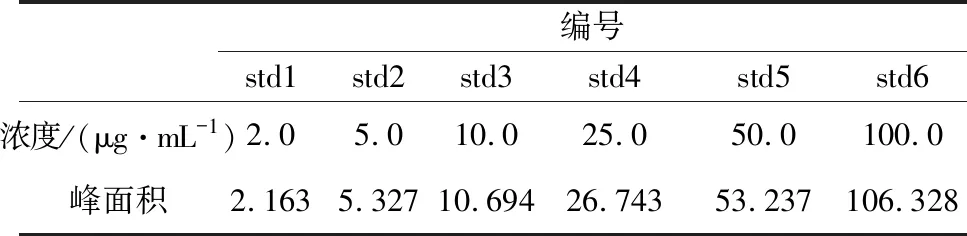
表1 線性關系結果(n=2)Tab 1 Linear relationship results(n=2)
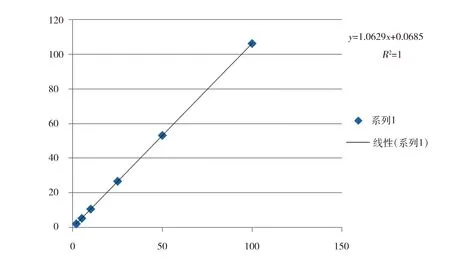
圖4 呋塞米含量測定標準曲線Fig 4 Standard curve of furosemide
2.2.4 檢測限 按1.3.2.2.5項下配制檢測限溶液進行測定,以光譜圖失真的最大濃度作為方法的檢測限,結果見圖5。結果表明恩諾沙星注射液制劑中呋塞米的檢測限為1.0 μg/mL。
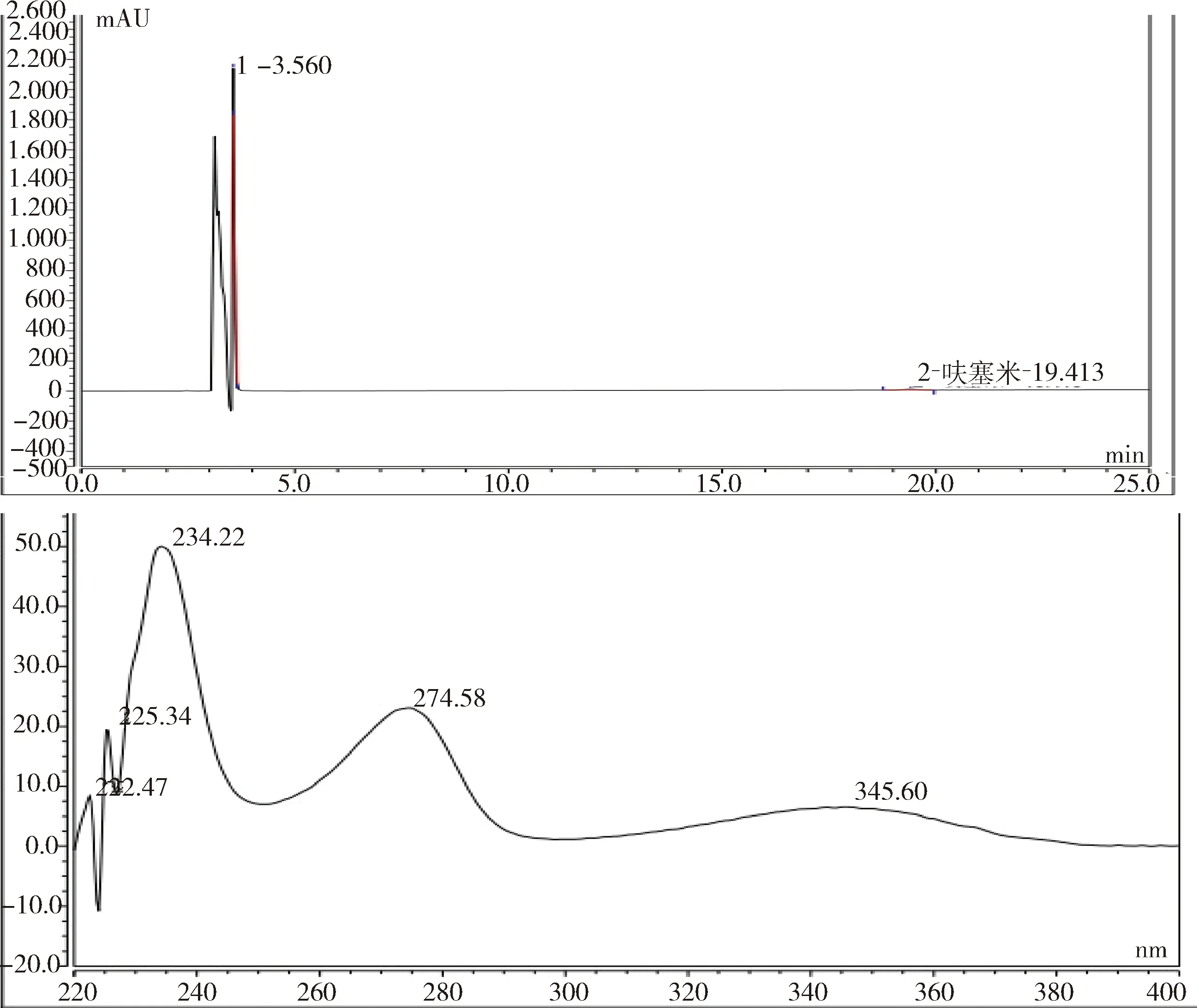
圖5 檢測限測定色譜光譜圖Fig 5 The chromatogram and spectrum of the LOD solution
2.2.5 準確度 通過對制劑空白樣品中添加呋塞米對照品進行回收率試驗,考察方法的準確度。按1.3.2.2.4項下每個濃度水平分別平行配制3份陽性模擬添加樣品溶液,進行測定,每個平行進樣2次,計算回收率,結果見表2。結果表明,制劑空白樣品中添加呋塞米的平均回收率為100.8%,RSD=1.0%。
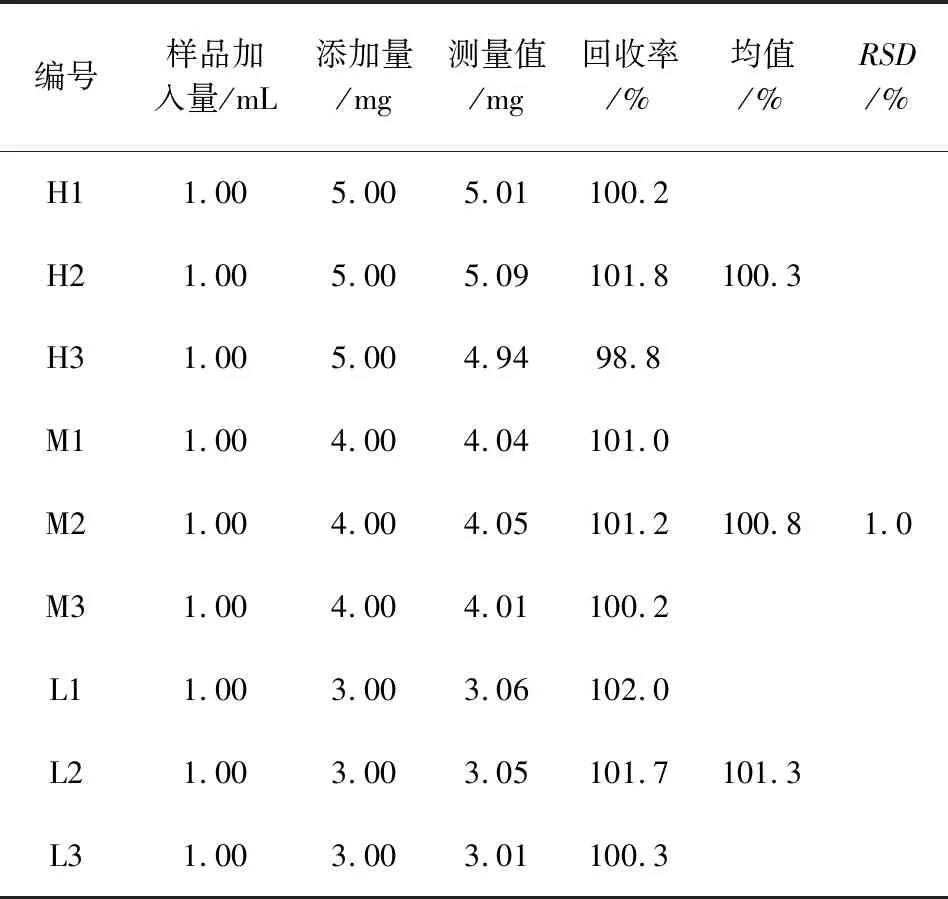
表2 回收率試驗結果Tab 2 Recovery test results
2.2.6 耐用性 以對照品溶液作為耐用性試驗溶液,通過改變柱溫、流速兩個方面考察方法的耐用性,結果見表3。試驗結果表明,當柱溫升高、流速增加時,保留時間會提前,同時,分離度均滿足檢測要求,說明方法耐用性良好。
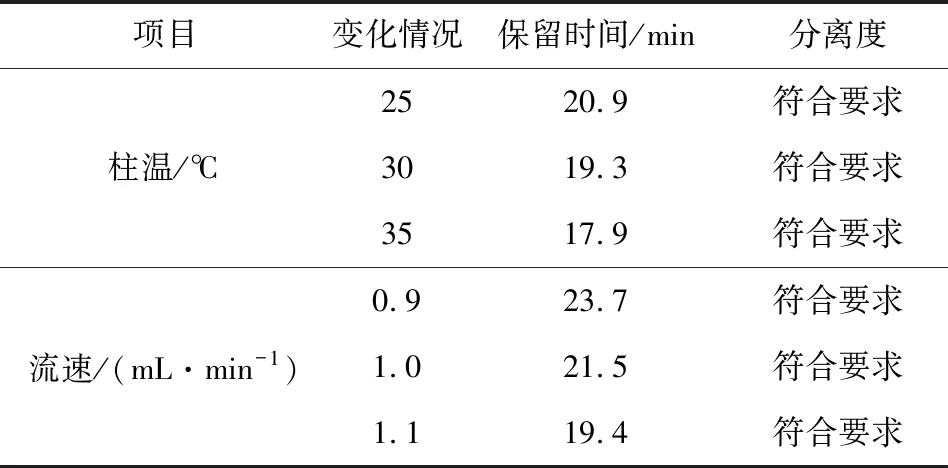
表3 耐用性試驗結果Tab 3 Durability test results
2.2.7 供試品測定 中國獸醫藥品監察所和貴州省獸藥飼料檢測所兩家單位根據擬定的恩諾沙星注射液中非法添加呋塞米檢查方法(1.3.1項)對供試品分別進行測定,均確認本批次恩諾沙星注射液(批號為20210501)中非法添加了呋塞米,并測得呋塞米的量分別為:4.8 g/L(中國獸醫藥品監察所),4.2 g/L(貴州省獸藥飼料檢測所),兩家單位結果基本一致。
3 討論與小結
3.1 溶媒和流動相的選擇 陳啟友[2]采用酸沉乙醚萃取分離恩諾沙星和呋塞米;儲蓉等[14]使用甲醇結合超聲處理提取功能食品中的呋塞米;《中國藥典》和《中國獸藥典》使用冰醋酸-乙腈-水(22∶489∶489)作為溶媒提取呋塞米。而采用高效液相色譜法測量呋塞米含量的流動相主要有“水-四氫呋喃-冰醋酸”和“水-甲醇-磷酸鹽緩沖液”兩種系統。為保持方法的延續性,選擇《中國藥典》和《中國獸藥典》使用的溶媒“冰醋酸-乙腈-水(22∶489∶489)”及流動相“水-四氫呋喃-冰醋酸(70∶30∶1)”作為方法的溶媒和流動相。
3.2 提取波長選擇 試驗色譜條件下,呋塞米對照品的光譜圖中有三個最大吸收波長,分別為236、274和344 nm。《中國藥典》和《中國獸藥典》中呋塞米注射液的含量測定波長均為272 nm。試驗獲得的274 nm和《中國藥典》及《中國獸藥典》中的272 nm雖然具有2 nm的偏差,但屬于允許的偏差范圍,故最終選擇272 nm作為方法的提取波長。
3.3 測量方法的選擇 查閱文獻可知,測量呋塞米的方法主要有可見-紫外分光光度法、高效液相色譜法和高效液相色譜串聯質譜法。可見-紫外分光光度法方法雖然簡單、快捷,但測量時需要分離出純凈的呋塞米,前處理方法較復雜、繁瑣;高效液相色譜串聯質譜法測量呋塞米的樣品前處理方法簡單,一般只需采用甲醇直接提取,但設備昂貴,要求操作人員須具備較高的技術水平;高效液相色譜法則相對簡單快捷,還可同時分離和測量多個復雜的組分,故選擇高效液相色譜法。
該方法準確可靠,可以作為恩諾沙星注射液非法添加呋塞米的檢查方法。
