基于化學(xué)計量學(xué)的三七傷藥片質(zhì)量控制方法研究
2023-11-25 07:54:51趙振霞雷蓉耿蓮朱靖劉永利
中國民族民間醫(yī)藥·上半月 2023年10期
趙振霞 雷蓉 耿蓮 朱靖 劉永利

【摘要】目的:建立三七傷藥片的TLC鑒別方法和HPLC含量測定方法,并結(jié)合化學(xué)計量學(xué)構(gòu)建其質(zhì)量控制體系。方法:采用TLC法對冰片、骨碎補和赤芍進(jìn)行鑒別,采用HPLC法對三七中5種皂苷類成分進(jìn)行含量測定。結(jié)合化學(xué)計量學(xué)方法進(jìn)行分析,以挖掘不同生產(chǎn)企業(yè)樣品間對質(zhì)量控制具有顯著貢獻(xiàn)的主要成分。結(jié)果:薄層色譜鑒別斑點清晰,分離度好,陰性無干擾;方法線性關(guān)系良好,r=0.9999,平均加樣回收率(n=9)分別為100.0%、99.9%、100.6%、99.0%和99.6%。通過化學(xué)計量學(xué)分析,尋找到2個對模型貢獻(xiàn)較大的成分,包括三七皂苷R1(VIP=1.2131)和人參皂苷Rd(VIP=1.1260),可進(jìn)一步深化三七傷藥片質(zhì)量控制研究。結(jié)論:所建立的方法專屬性強、準(zhǔn)確度高、重復(fù)性好,化學(xué)計量學(xué)分析為三七傷藥片質(zhì)量控制的進(jìn)一步研究提供了重要參考。
【關(guān)鍵詞】三七傷藥片;質(zhì)量控制;薄層色譜;高效液相色譜;化學(xué)計量學(xué)
【中圖分類號】R284.1【文獻(xiàn)標(biāo)志碼】 A【文章編號】1007-8517(2023)19-0030-08
DOI:10.3969/j.issn.1007-8517.2023.19.zgmzmjyyzz202319007
The Research of Quality Control for Sanqishangyao Tablets Based on ChemometricsZHAO Zhenxia1LEI Rong1GENG Lian1ZHUJing1,2LIU Yongli1*
1.Hebei Institute for Drug and Medical Device Control,HEBEI Key Laboratory of Traditional Chinese Medicine
Quality Evaluation And Standard Research, Shijiazhuang 050227,China;
2 .Hebei Medical University, Shijiazhuang 050017,ChinaAbstract:Objective To establish TLC identification method and HPLC determination method for Sanqishangyao Tablets and build its quality control system combined with chemometrics. Methods Borneolumsyntheticum,Drynariae Rhizoma and Paeoniae Radix Rubra were identified by TLC. The contents of five saponins in Notoginseng Radix et Rhizoma were determined by HPLC. Combined with the chemometrics methods, the main components of samples from different production enterprise which had significant contribution to the quality control were analyzed. Results The spots on the TLC plate were clear, the resolution was well and without interference for negative control sample. The linear relationships of calibration curves were favorable,r=0.9999.The recoveries(n=9)of notoginsenoside R1,ginsenoside Rg1,ginsenoside Re,ginsenoside Rb1, ginsenoside Rd were 100.0%、99.9%、100.6%、99.0%和99.6%.Through chemometrics analysis, it was found that 2 component scontributed significantly to the model, including notoginsenoside R1(VIP=1.2131) and ginsenoside Rd (VIP=1.1260), which can further deepen the quality control research of SanqishangyaoTablets. Conclusion The methods of established were specificity,accuracy and well repeatability. Further through the chemometrics, which provided an important reference for the quality control research of Sanqishangyao Tablets.
Keywords:Sanqishangyao Tablets;Quality Control;Thin Layer Chromatography;High Performance Liquid Chromatography;Chemometrics
三七傷藥片由三七、制草烏、雪上一枝蒿、冰片、骨碎補、紅花、接骨木、赤芍等組成,具有舒筋活血,散瘀止痛功效,是臨床骨傷科疾病中一種常用的中成藥,用于治療跌打損傷、風(fēng)濕瘀阻、關(guān)節(jié)痹痛,急慢性扭傷損傷等[1]。現(xiàn)代藥理學(xué)研究[2-3]表明三七傷藥片還具有鎮(zhèn)痛、抗炎,改善血液流變性的作用,因此在臨床上更是得到了廣泛應(yīng)用。為控制藥品質(zhì)量及提高臨床用藥的安全性,本文對三七傷藥片中主要藥味的質(zhì)量控制方法進(jìn)行研究。該品種收載于《中國藥典》2020年版一部,鑒別項僅收載了三七中三七皂苷R1、人參皂苷Rg1和赤芍中芍藥苷的薄層色譜鑒別,對方中藥味控制較少,且未有對處方中君藥三七進(jìn)行含量測定,難以全面控制其內(nèi)在質(zhì)量[4],因此,研究參照相關(guān)文獻(xiàn)[5-8]建立了三七傷藥片中3種藥味的薄層色譜鑒別方法,并建立了三七中五種皂苷類成分的含量測定方法,再結(jié)合化學(xué)計量學(xué)分析影響三七傷藥片質(zhì)量的主要因素,為后續(xù)三七傷藥片質(zhì)量標(biāo)準(zhǔn)的建立提供可靠的技術(shù)保障。
1儀器與材料
1.1儀器Ultimate 3000高效液相色譜儀(包括四元泵和紫外檢測器,美國賽默飛公司);KQ-800KDE型超聲波清洗器(昆山市超聲儀器有限公司);SYG-1-6數(shù)顯恒溫水浴鍋(天津市泰斯特儀器有限公司);FED56電熱恒溫干燥箱(德國賓德公司);Mettler XPE26電子天平(瑞士梅特勒公司,0.001 mg)、Mettler XS105電子天平(瑞士梅特勒公司,0.01 mg);Milli-Q 型超純水凈化系統(tǒng)(美國 Millipore 公司)。
1.2材料冰片(批號:110713-201706)、骨碎補(批號:121169-202105)、柚皮苷(批號:110722-201714)、赤芍(批號:121093-201804)、芍藥苷(批號:110736-202145)、三七皂苷R1(批號:110745-201921)、人參皂苷Rg1(批號:110703-20235)、人參皂苷Re(批號:110754-202104)、人參皂苷Rb1(批號:110704-202230)、人參皂苷Rd(批號:111818-201302),上述對照品及對照藥材均購買于中國食品藥品檢定研究院;17批次三七傷藥片樣品購自藥店,詳細(xì)信息見表1;乙腈(色譜純,德國Merck公司),水為超純水;其余試劑均為分析純。
2方法與結(jié)果
2.1鑒別研究
2.1.1冰片的TLC鑒別取不同生產(chǎn)企業(yè)的三七傷藥片樣品三批(批號:20190813、191003、20191224)各40片,采用刮片法除去外層糖衣,置研缽內(nèi)研細(xì)后轉(zhuǎn)移至150 mL錐形瓶中,加入試劑乙醚20 mL,蓋緊瓶塞,室溫放置20 min后,超聲處理(功率為250 W,頻率為40 kHz)10 min,過濾,濾液低溫?fù)]干,殘渣加三氯甲烷1 mL使其溶解,作為樣品溶液。按《中國藥典》2020年版一部三七傷藥片標(biāo)準(zhǔn)中【處方】比例與【制法】項下制備工藝,制備缺冰片藥味的陰性樣品,按“樣品溶液”項下方法制備陰性樣品溶液。取冰片對照品1.052 mg,加三氯甲烷1 mL使其溶解,作為對照品溶液。吸取上述制得的三批樣品溶液、陰性樣品溶液及對照品溶液各5 μL,分別點于同一塊硅膠G薄層板上,以甲苯-乙酸乙酯(19∶1)為展開劑,展開,取出,晾干,噴以顯色劑1%香草醛硫酸溶液,在105 ℃加熱至斑點顯色清晰。結(jié)果:樣品色譜中,在與冰片對照品色譜相應(yīng)的位置上,顯相同顏色的斑點,且斑點清晰、分離度好,陰性樣品無干擾(如圖1所示),說明方法可行、專屬性良好。
2.1.2骨碎補的TLC鑒別取不同生產(chǎn)企業(yè)的三七傷藥片樣品三批(批號:20190813、191003、20191224)各15片,采用刮片法除去外層糖衣,置研缽內(nèi)研細(xì)后轉(zhuǎn)移至150 mL錐形瓶中,加入試劑甲醇50 mL,82 ℃水浴條件下加熱回流提取1 h,過濾,將濾液蒸干后,殘渣加入水溶液20 mL使其溶解,然后用水飽和的正丁醇溶液振搖提取3次,每次用量均為25 mL,合并正丁醇提取液,再用正丁醇飽和的水洗滌2次,每次用量均為25 mL,正丁醇液置100 ℃水浴鍋上,蒸干,殘渣加入甲醇1 mL使其溶解,作為樣品溶液。按《中國藥典》2020年版一部三七傷藥片標(biāo)準(zhǔn)中【處方】比例與【制法】項下制備工藝,制備缺骨碎補藥味的陰性樣品,按照“樣品溶液”項下方法制備陰性樣品溶液。取骨碎補對照藥材2.0235 g,置150 mL錐形瓶中,加入乙醇15 mL,超聲處理(功率為250 W,頻率為40 kHz)20 min,過濾,濾液置100 ℃水浴鍋上,蒸干,殘渣加入甲醇1 mL使其溶解,作為對照藥材溶液。另取柚皮苷對照品1.357 mg,加甲醇1 mL使其溶解,作為對照品溶液。照薄層色譜法試驗,吸取上述制得的三批樣品溶液和陰性樣品溶液各10 μL、骨碎補對照藥材和柚皮苷對照品溶液各5 μL,分別點于同一塊硅膠G薄層板上,以甲苯-乙酸乙酯-甲酸-水(1∶12∶2.5∶3)的上層溶液為展開劑,展開,取出,晾干,噴以顯色劑三氯化鋁試液,熱風(fēng)吹干后,置紫外光燈(365 nm)下檢視。結(jié)果:樣品色譜中,在與骨碎補對照藥材和柚皮苷對照品色譜相應(yīng)的位置上,顯相同顏色的斑點,且斑點清晰、分離度好,陰性樣品無干擾(如圖2所示),說明方法可行、專屬性良好。
2.1.3赤芍的TLC鑒別同骨碎補的TLC鑒別制備樣品溶液。按《中國藥典》2020年版一部三七傷藥片標(biāo)準(zhǔn)中【處方】比例與【制法】項下制備工藝,制備缺赤芍藥味的陰性樣品,按“樣品溶液”項下方法制備陰性樣品溶液。取赤芍對照藥材1.0827 g,置150 mL錐形瓶中,加入試劑乙醇15 mL,超聲處理(功率為250 W,頻率為40 kHz)20 min,過濾,濾液蒸干,殘渣加甲醇1 mL使其溶解,作為對照藥材溶液。再取芍藥苷對照品1.079 mg,加甲醇1 mL使其溶解,作為對照品溶液。照薄層色譜法試驗,吸取三批樣品溶液和陰性樣品溶液各5 μL、赤芍對照藥材和芍藥苷對照品溶液各2 μL,分別點于同一塊硅膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40∶5∶10∶0.2)為展開劑,展開,取出,晾干,噴以顯色劑5%香草醛硫酸溶液,在105 ℃加熱至斑點顯色清晰。結(jié)果:樣品色譜中,在與赤芍對照藥材色譜和芍藥苷對照品色譜相應(yīng)的位置上,顯相同顏色的斑點,且斑點清晰、分離度好,陰性樣品無干擾(如圖3所示),說明方法可行、專屬性良好。
2.2三七中5種成分含量測定
2.2.1色譜條件采用資生堂MGII C18(4.6 mm×250 mm,5 μm)色譜柱,流動相:以乙腈為流動相A,水為流動相B。梯度洗脫:0~15 min,A保持20%;15~35? min,A 20%→22%;35~50 min,A 22%→24%;50~110 min,A 24%→40%,流速為1.0 mL/min,柱溫為25 ℃,檢測波長為203 nm。
2.2.2溶液的制備
2.2.2.1混合對照品溶液取待測五種皂苷類對照品,三七皂苷R17.529 mg、人參皂苷Rg125.306 mg、人參皂苷Re 5.145 mg、人參皂苷Rb1 25.179 mg和人參皂苷Rd 7.504 mg置同一25 mL量瓶中,加入甲醇約20 mL超聲處理(功率為250 W,頻率為40 kHz)10 min使溶解,取出,放冷至室溫后,用甲醇稀釋并定容至刻度,搖勻,即得混合對照品溶液。
2.2.2.2樣品溶液取三七傷藥片樣品(批號:20181202)20片,采用刮片法除去外層糖衣,精密稱定重量后,置研缽內(nèi)研細(xì),取3.0045 g,置150 mL錐形瓶中,精密加入甲醇提取液50 mL,稱定重量,82 ℃水浴條件下加熱回流提取1 h,放冷,再稱定重量,用甲醇補足回流過程中損失的重量,搖勻,過濾,精密量取續(xù)濾液25 mL置蒸發(fā)皿中,水浴100 ℃條件下蒸干,殘渣加入水溶液25 mL使其溶解,用乙酸乙酯振搖提取2次,每次用量均為20 mL,棄去乙酸乙酯液,水液繼續(xù)用水飽和的正丁醇溶液振搖提取4次,每次用量均為25 mL,合并正丁醇提取液,水浴100 ℃條件下蒸干,殘渣加甲醇使其溶解并轉(zhuǎn)移至5 mL量瓶中,加甲醇稀釋至刻度,搖勻,過濾,取續(xù)濾液,即得樣品溶液。
2.2.2.3陰性樣品溶液按《中國藥典》三七傷藥片標(biāo)準(zhǔn)中【處方】比例與【制法】項下制備工藝,制備缺三七藥味的陰性樣品,按“樣品溶液”項下方法制備陰性樣品溶液。
2.2.3專屬性試驗取混合對照品溶液、樣品溶液及陰性樣品溶液,按照“2.2.1”項下的色譜條件,分別取10 μL注入高效液相色譜儀,進(jìn)行分析,并記錄色譜圖,如圖4所示。在樣品色譜圖中,分別顯示與5種待測對照品保留時間一致的色譜峰,各色譜峰之間分離度良好,陰性樣品色譜圖中無相應(yīng)的色譜峰,說明方法可行,且除三七外的其它藥味不干擾三七傷藥片中待測成分的測定,方法專屬性良好。
2.2.4線性關(guān)系考察精密稱取三七皂苷R1 31.18 mg、人參皂苷Rg1 89.98 mg、人參皂苷Re 23.40 mg、人參皂苷Rb1 91.08 mg及人參皂苷Rd 32.42 mg置同一10 mL量瓶中,加入甲醇約8 mL,超聲處理(功率為250 W,頻率為40 kHz)15 min使溶解,取出,放冷至室溫后,用甲醇稀釋并定容至刻度,搖勻,即得混合對照品儲備液。精密吸取上述對照品儲備液0.1 mL、0.2 mL、0.5 mL、1.0 mL、5.0 mL,分別置于5個10 mL量瓶中,加甲醇稀釋并定容至刻度,搖勻,即得系列濃度的混合對照品溶液。精密吸取上述系列溶液各10 μL,分別注入高效液相色譜儀,按照“2.2.1”項下的色譜條件測定,以對照品進(jìn)樣量(μg)為橫坐標(biāo)(x),峰面積積分值為縱坐標(biāo)(y),繪制標(biāo)準(zhǔn)曲線,求得回歸方程。結(jié)果見表2。
2.2.5穩(wěn)定性試驗取按照“2.2.2”項下方法制備的批號為20181202的樣品溶液,分別在配制后0 h、3 h、7 h、13 h、19 h、24 h按照“2.2.1”項下色譜條件進(jìn)樣測定,記錄上述6個時間點5種成分色譜峰峰面積積分值,并計算各成分的RSD值,結(jié)果三七皂苷R1、人參皂苷Rg1、人參皂苷Re、人參皂苷Rb1和人參皂苷Rd的RSD值分別為1.2%、1.0%、0.9%、0.4%、1.0%,結(jié)果表明本實驗制得的三七傷藥片供試品溶液在24 h內(nèi)穩(wěn)定。
2.2.6重復(fù)性試驗取同一批樣品(批號:20181202),采用刮片法除去外層糖衣,置研缽內(nèi)研細(xì),取1.5 g、3.0 g、4.5 g各三份,精密稱定每份重量,按照“2.2.2”項下方法分別制備樣品溶液,按照“2.2.1”項下色譜條件進(jìn)樣測定,記錄每一份樣品中各成分峰面積積分值,計算5種皂苷類成分的含量及其RSD值,詳細(xì)結(jié)果見表3,表明本實驗建立的方法重復(fù)性良好。
2.2.7加樣回收率試驗精密稱取經(jīng)重復(fù)性試驗測定,含量已知的三七傷藥片樣品(批號:20181202)粉末9份,每份重量約1.5 g,每三份分為一組,分別加入用甲醇配制的低、中、高三個濃度的混合對照品溶液,按照“2.2.2”項下方法分別制備回收率測定用樣品溶液,按照“2.2.1”項下色譜條件進(jìn)樣測定,計算每份樣品中待測成分回收率及其平均值和RSD值,詳細(xì)結(jié)果見表4,表明所建立的方法回收率良好。
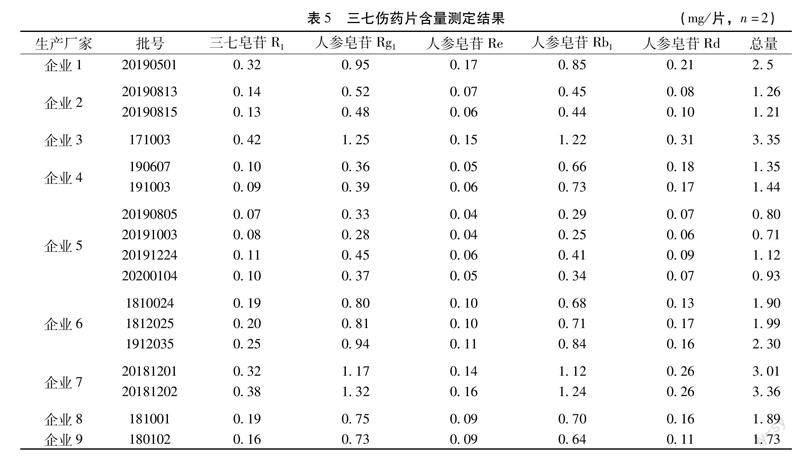
2.3樣品測定取收集到的17批三七傷藥片樣品,按上述建立的方法進(jìn)行試驗,結(jié)果顯示17批樣品,3個TLC鑒別均有相應(yīng)的斑點檢出,5種皂苷成分含量測定結(jié)果詳見表5。
2.4基于化學(xué)計量學(xué)的三七傷藥片質(zhì)量分析從以上的樣品檢驗結(jié)果可看出,收集到的17批樣品,3個薄層鑒別均有相應(yīng)的斑點檢出,全部符合規(guī)定,從鑒別的角度尚不能對樣品的品質(zhì)優(yōu)劣做出區(qū)分,于是針對三七中皂苷類成分的含量進(jìn)行了化學(xué)計量學(xué)的相關(guān)分析。
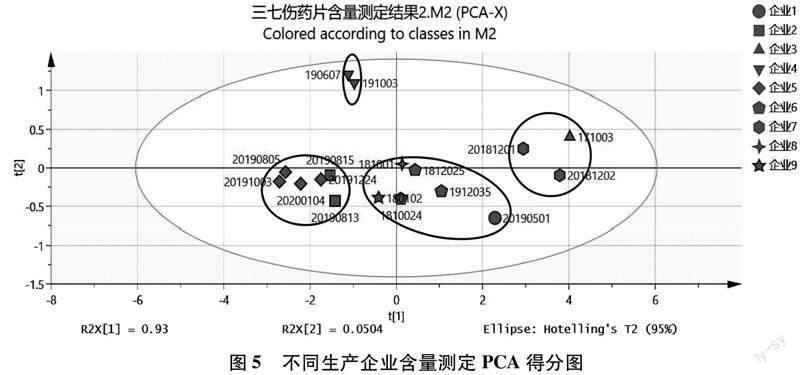
2.4.1主成分分析(PCA)將9個生產(chǎn)企業(yè)生產(chǎn)的共計17批次三七傷藥片樣品中五種皂苷成分的含量測定結(jié)果導(dǎo)入SIMCA 14.1軟件進(jìn)行PCA分析,所有的數(shù)據(jù)都落在95%置信區(qū)間,且累積方差貢獻(xiàn)率R2X[1]與R2X[2]之和大于80%,說明所擬合的PCA得分圖有很好的參考性。17批樣品的二維PCA得分圖見圖5,從PCA得分圖可看出,17批樣品大致被分為了4類,同一企業(yè)的樣品基本都可聚到一類,但企業(yè)間樣品差距較大,企業(yè)3和企業(yè)7的樣品中皂苷成分的含量較高,聚為一類;而企業(yè)2和企業(yè)5的樣品含量較低,同樣聚為一類。
2.4.2正交偏最小二乘法-判別分析(OPLS-DA)為了進(jìn)一步尋找三七傷藥片的差異質(zhì)量標(biāo)志物,以皂苷類成分的含量為變量,使用數(shù)據(jù)統(tǒng)計軟件SIMCA 14.1對17批樣品進(jìn)行OPLS-DA分析,建立OPLS-DA模型,模型得分圖如圖6所示。由圖6可見,在95%的置信區(qū)間內(nèi),OPLS-DA能有效的將樣品進(jìn)行區(qū)分,且與PCA的分類結(jié)果基本一致。OPLS-DA模型中變量重要性投影值(variable importance for the projection,VIP值)可直觀反映出具有統(tǒng)計學(xué)意義的差異質(zhì)量標(biāo)志物,根據(jù)VIP>1.0的原則,篩選差異組分,結(jié)果共找到2個成分,按照VIP值大小依次為三七皂苷R1(1.2131)>人參皂苷Rd(1.1260)。如圖7所示。
3討論
3.1薄層鑒別方法研究參考2020年版《中國藥典》一部及相關(guān)文獻(xiàn)[9-11],對組方中各味藥進(jìn)行TLC鑒別方法的探索,結(jié)果三七、紅花等藥味的鑒別方法因分離度較差、斑點不清晰等原因暫未列入質(zhì)量標(biāo)準(zhǔn);制草烏、雪上一枝蒿為毒性藥味,需對其進(jìn)行嚴(yán)格控制,既要對毒性較大的雙酯型生物堿進(jìn)行限量檢查,又要對主要的藥效成分單酯型生物堿進(jìn)行含量測定,后續(xù)對其進(jìn)行了HPLC方法的一系列研究工作。本文采用TLC法建立了方中冰片、骨碎補、接骨木、赤芍3味藥的薄層色譜鑒別方法,經(jīng)過多次試驗選定薄層鑒別條件,結(jié)果方法簡便易行,斑點清晰圓整,分離度好,陰性無干擾,試驗過程中筆者還對多個廠家生產(chǎn)的薄層板(青島海洋化工廠、煙臺化學(xué)工業(yè)研究所、德國Merck公司)進(jìn)行了考察,結(jié)果均能很好地分離,表明所建立方法有較好的耐用性,可作為三七傷藥片的質(zhì)量控制方法。
3.2流動相及供試品溶液制備方法的選擇由于本品為復(fù)方制劑,成分復(fù)雜,為使三七皂苷R1、人參皂苷Rg1、人參皂苷Re、人參皂苷Rb1和人參皂苷Rd各色譜峰分離度達(dá)到要求,且峰形良好,本實驗參照相關(guān)文獻(xiàn)[12-15]對色譜條件及樣品溶液的制備方法都進(jìn)行了優(yōu)化。預(yù)實驗中分別以乙腈-水、乙腈-0.1%磷酸水溶液、乙腈-0.1%冰醋酸、甲醇-水、甲醇-0.1%磷酸水溶液(梯度洗脫)作為流動相進(jìn)行試驗,結(jié)果采用乙腈-水溶液時,色譜峰分離效果好,峰形較佳;在提取方式的選擇上,比較了直接提取、正丁醇萃取凈化處理、大孔吸附樹脂柱凈化處理的方法,結(jié)果采用正丁醇萃取凈化處理的方法,可較大程度去除雜質(zhì)峰的干擾;選擇供試品溶液提取溶劑時,將50%甲醇、70%甲醇、甲醇、乙醇作對比,結(jié)果甲醇提取效率最高。所建立的方法,被測成分色譜峰峰形良好,相互之間無干擾,具有重現(xiàn)性好、靈敏度高、選擇性強、耐用性好等特點,是控制三七傷藥片質(zhì)量的有效方法。
3.3含量測定結(jié)果分析本文采用HPLC法測定三七傷藥片制劑中5種皂苷類成分的含量,通過化學(xué)計量學(xué)的分析可知,同一廠家生產(chǎn)的不同批次樣品中,5種成分含量基本相當(dāng),一致性較好,而不同廠家生產(chǎn)的樣品含量差異較大,且三七中的專屬性成分三七皂苷R1是影響其質(zhì)量的關(guān)鍵因素,因此亟需建立三七傷藥片中君藥三七的含量測定方法,且指標(biāo)成分的選擇不能僅限于五加科植物中共有的皂苷類成分人參皂苷Rg1、人參皂苷Re和人參皂苷Rb1等,而必須對三七的專屬性成分三七皂苷R1加以控制,以確保其臨床療效的穩(wěn)定。產(chǎn)品的質(zhì)量與所用藥材質(zhì)量、生產(chǎn)工藝等都有密切關(guān)系,要提高產(chǎn)品質(zhì)量,不但要監(jiān)控原料的質(zhì)量,而且還要對生產(chǎn)工藝進(jìn)行優(yōu)化、確保質(zhì)量的穩(wěn)定均一,所以建立能真正表征藥品品質(zhì)的質(zhì)量標(biāo)準(zhǔn)顯得尤為重要[16],質(zhì)量控制體系的建立應(yīng)以體現(xiàn)產(chǎn)品的質(zhì)量為核心。
綜上所述,研究同時建立了三七傷藥片中3味藥的薄層色譜鑒別方法及三七多指標(biāo)成分的含量測定方法,建立的質(zhì)量控制方法涉及藥味多,質(zhì)控成分種類多,能相對全面地控制三七傷藥片的質(zhì)量,保證臨床用藥的安全性與有效性。本次試驗過程中,筆者同時對系列品種三七傷藥膠囊與三七傷藥顆粒進(jìn)行了研究,對該系列制劑通用標(biāo)準(zhǔn)的建立具有指導(dǎo)意義,同時對全面控制產(chǎn)品質(zhì)量的穩(wěn)定性和臨床療效的一致性具有重要意義。參考文獻(xiàn)
[1]國家藥典委員會.中華人民共和國藥典(一部) [S].北京:中國醫(yī)藥科技出版社, 2020: 505-506.
[2]喻林華,曾獻(xiàn),王元清,等.三七傷藥片的抗炎消腫藥理作用研究[J]. 中國實用醫(yī)藥,2007,2(32): 16-18.
[3]喻林華,王元清,曾獻(xiàn),等. 三七傷藥片的抗凝血藥理作用研究[J]. 中醫(yī)藥導(dǎo)報,2007,13(9): 19-20.
[4]高海燕,王一名,李娜. 價格與成本倒掛專項抽驗對三七傷藥片中“三七”的質(zhì)量分析[J]. 抗感染藥學(xué),2018,15(3): 383-385.
[5]諶順清,梁偉,張雪妹,等.骨碎補化學(xué)成分和藥理作用研究進(jìn)展[J]. 中國中藥雜志,2021,46(11): 2737-2745.
[6]黃依丹,成嘉欣,石穎,等.近五年三七化學(xué)成分、色譜分析、三七提取物和藥理活性的研究進(jìn)展[J]. 中國中藥雜志, 2022,47(10): 2584-2596.
[7]張桂英,許崇德,劉建芳,等. HPLC 法測定消栓通絡(luò)片中3 種三七成分的含量[J]. 藥學(xué)研究,2021,40(9): 578-581.
[8]趙振霞,王敏,段吉平,等.三七葉、三七花質(zhì)量標(biāo)準(zhǔn)研究[J]. 中國現(xiàn)代中藥,2018,20(8): 979-983.
[9]蔣亞奇,高天陽,張良雨,等. 三七的薄層鑒別研究[J].時珍國醫(yī)國藥,2020,31(2): 354-355.
[10]周慧慧,杜少兵,高速,等. 丹參-紅花配方顆粒制備工藝的優(yōu)化、TLC 鑒別及含量測定[J].華西藥學(xué)雜志,2022,37(3): 292-296.
[11]何風(fēng)艷,何軼,戴忠,等. HPLC-MS/MS 法同時測定骨刺片中7 個生物堿的含量[J]. 藥物分析雜志,2018,38(10): 1810-1816.
[12]樊紅軍,谷杰,周麗麗,等. HPLC 法測定三七傷藥片、膠囊和顆粒中人參皂苷Rg1、人參皂苷Rb1的含量[J]. 中國衛(wèi)生產(chǎn)業(yè),2017,13(5): 77-83.
[13]梁笑,錢珊珊,孫寒,等.《中國藥典》三七項下供試品溶液制備方法的改進(jìn)[J]. 中草藥,2019,50(17): 4145-4151.
[14]梁洪,李波,劉艷芳,等. HPLC法測定田參氨基酸膠囊中三七皂苷R1、人參皂苷Rg1、Re、Rb1、Rd含量[J]. 今日藥學(xué),2017,27(8): 528-531.
[15]蔣騰川,王艷林,洪影雯,等. 不同生長年限對三七主根中總皂苷及浸出物含量的影響[J].廣西中醫(yī)藥,2022,45(2): 59-63.
[16]馬雙成,王瑩,魏鋒. 中藥質(zhì)量控制未來發(fā)展方向的思考[J].中國藥學(xué)雜志,2021,56(16): 1273-1281.
(收稿日期:2023-01-12編輯:陶希睿)
猜你喜歡
熱帶農(nóng)業(yè)科學(xué)(2016年10期)2016-12-12 01:52:56
分析化學(xué)(2016年7期)2016-12-08 00:57:07
中國科技博覽(2016年18期)2016-10-19 11:09:28
中國科技博覽(2016年18期)2016-10-19 09:03:36
中國科技博覽(2016年18期)2016-10-19 08:46:18
科技視界(2016年21期)2016-10-17 17:58:28
科學(xué)與財富(2016年28期)2016-10-14 04:01:52
科技視界(2016年20期)2016-09-29 13:11:33
科技視界(2016年20期)2016-09-29 13:10:51
科技視界(2016年20期)2016-09-29 13:10:08
