
基于HPLC梯度洗脫-切換波長法測定防芷鼻炎片的6種成分
2024-01-08 08:41:24余清蓮
中國民族民間醫藥 2023年23期
余清蓮
福建省三明市檢驗檢測中心,福建 三明 365000
防芷鼻炎片由蒼耳子、野菊花、鵝不食草等十味中藥制成,用于治療慢性鼻炎引起的噴嚏、鼻塞、頭痛、過敏性鼻炎、慢性鼻竇[1]。防芷鼻炎片的現行質量標準[2]對性狀、化學鑒別做出了相關規定,相關文獻主要對蒼耳子、白芷、墨旱蓮、野菊花、白芍及防風的薄層色譜鑒別[3-5]、HPLC測定芍藥苷的含量[6]、HPLC測定歐前胡素、異歐前胡素的含量[7]及RRLC測定蒙花苷、芍藥苷的含量[8]等方面展開研究。防芷鼻炎片組方復雜,僅是鑒別、檢查、單個成分或兩個成分的含量測定難以全面反應該藥的質量。因此,實驗基于中醫方劑配伍組成的基本原則,選取君藥蒼耳子的質量標志物的綠原酸、咖啡酸[9],臣藥野菊花的質量標志物蒙花苷[10],臣藥白芍的質量標志物芍藥苷,佐藥防風藥效的質量標志物升麻素苷、5-O-甲基維斯阿米醇苷[11]為指標性成分,探索同時測定上述指標成分的含量測定方法,以期為更全面地控制防芷鼻炎片的質量提供科學參考。
1 儀器與試劑
1.1 儀器 1260高效液相色譜儀(安捷倫公司);HH-6數顯恒溫水浴鍋(常州榮華儀器制造有限公司);CPA324S電子分析天平(賽多利斯科學儀器公司)。
1.2 材料 綠原酸 (批號:110831-201906,含量91.5%)、咖啡酸 (批號:110753-201716,含量99.3%)、芍藥苷 (批號:110736-201943,含量95.1%)、升麻素苷 (批號110885-201703)、5-O-甲基維斯阿米醇苷(批號:11924-201806,含量99.5%)、蒙花苷(批號:11924-201806,含量99.5%)。甲醇(色譜純、分析純)、甲酸(分析純)、醋酸(分析純)。
2 方法與結果
2.1 色譜條件 色譜柱:unitary C18柱 (4.6 mm×150 mm,5 μm);流動相:甲醇(A)-0.5%醋酸溶液(B);梯度洗脫(0~10 min:20%A;10~40 min:20%A→35%A;40~55 min:35%A→45%A;55~68 min:45%A→55%A);流速:1 mL/min;進樣體積:10 μL;波長:0~21 min 323 nm、21~30 min 230 nm、30~60 min 254 nm、60 min后 330 nm。
2.2 溶液的制備
2.2.1 供試品溶液的制備 取防芷鼻炎片20片,除去糖衣,稱取重量后(平均片重為2.993 g),研磨成細粉,取0.6 g,精密加入甲酸-50%甲醇(1∶20)混合溶液25 mL,稱定重量,加熱回流2 h,放冷,稱重,用甲酸-50%甲醇(1∶20)混合溶液補足失重,濾過,取續濾液,即得。
2.2.2 混合對照品溶液的制備 精密稱取綠原酸12.86 mg、咖啡酸12.91 mg、芍藥苷12.6 mg、升麻素苷15.27 mg、5-O-甲基維斯阿米醇苷10.08 mg、蒙花苷9.51 mg,分別加入甲醇50 mL,配成對照品貯備液。取上述貯備液適量,加甲醇稀釋成每mL含綠原酸10.29 μg、咖啡酸3.1 μg、芍藥苷100.8 μg、升麻素苷12.216 μg、5-O-甲基維斯阿米醇苷4.032 μg、蒙花苷20.76 μg的混合對照品溶液。
2.3 方法學考察
2.3.1 系統適用性試驗[11]精密吸取混合對照品溶液、供試品溶液各10 μL,按“2.1”項的條件檢測,測定結果如圖1、圖2所示。在供試品溶液的色譜圖上,有與對照品保留時間一致的色譜峰,且色譜報告中顯示:各色譜峰的理論塔板數高于6500,分離度高于1.5,拖尾因子在0.95~1.05之間。表明該方法的系統適用性良好。
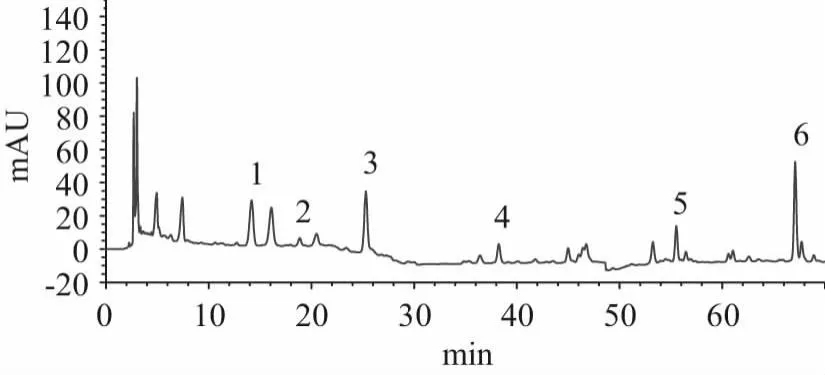
圖1 供試品溶液色譜圖
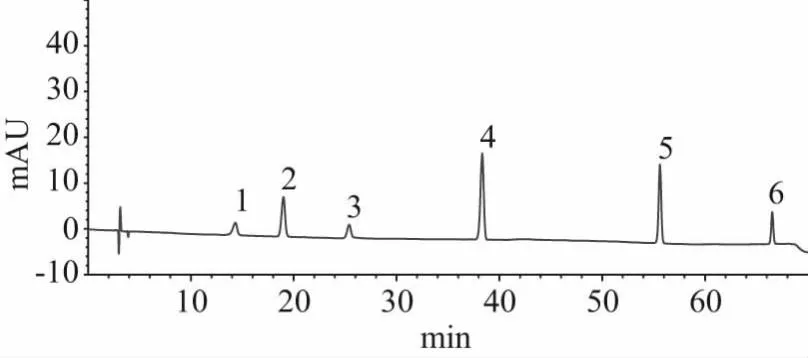
圖2 對照品溶液色譜圖
2.3.2 線性關系 精密吸取混合對照品溶液3 μL、6 μL、9 μL、12 μL、15 μL,分別按“2.1”項色譜條件進樣測定,以對照品的含有量及相應的峰面積為橫、縱坐標,繪制咖啡酸、綠原酸、芍藥苷、升麻素苷、5-O-甲基維斯阿米醇苷、蒙花苷的標準曲線并進行線性回歸。回歸方程見表1。
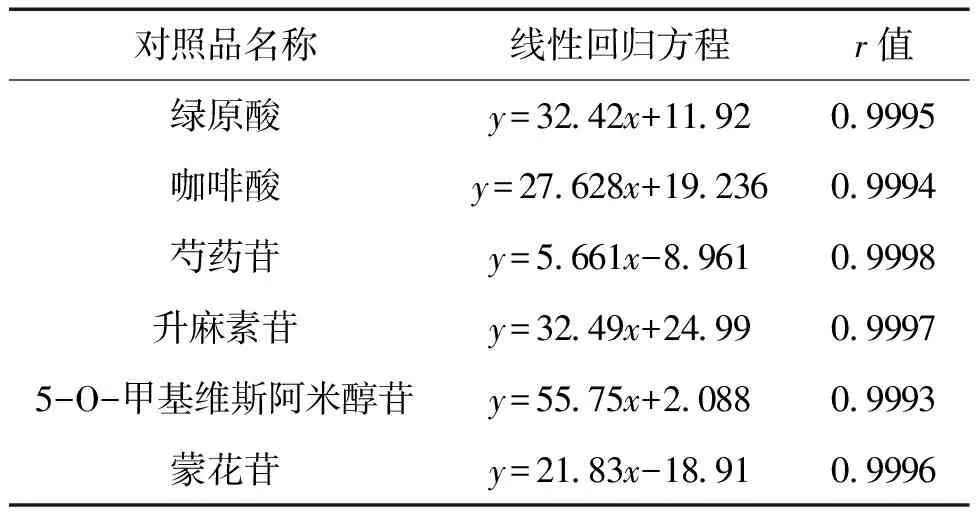
表1 線性回歸方程
2.3.3 穩定性試驗 分別在0 h、4 h、8 h、12 h、16 h、24 h精密吸取2.2.1項供試液10 μL,進樣測定,綠原酸、咖啡酸、芍藥苷、升麻素苷、5-O-甲基維斯阿米醇苷、蒙花苷峰面積的RSD分別為1.5%、2.1%、0.9%、1.6%、1.2%、1.8%,供試品溶液24 h內含量穩定。
2.3.4 重復性試驗 精密稱取防芷鼻炎片細粉6份,每份0.6 g,按“2.2.1”項方法制備6份供試品溶液,在“2.1”色譜條件下測定,每片含綠原酸、咖啡酸、芍藥苷、升麻素苷、5-O-甲基維斯阿米醇苷、蒙花苷量的平均值(RSD)分別為0.27 mg(1.2%)、0.04 mg(2.0%)、2.1 mg(1.0%)、0.13 mg(1.2%)、0.20 mg(1.6%)、0.61 mg(0.8%)。
2.3.5 精密度試驗 連續吸取“2.2.1”項供試品溶6次,每次10 μL,在“2.1”色譜條件下測定,綠原酸、咖啡酸、芍藥苷、升麻素苷、5-O-甲基維斯阿米醇苷、蒙花苷峰面積的RSD分別為1.0%、1.5%、0.9%、1.0%、0.8%、1.5%。
2.3.6 回收率試驗 精密稱取已知含量的防芷鼻炎片細粉6份,每份0.3 g,分別精密加入適量的綠原酸、咖啡酸、芍藥苷、升麻素苷、5-O-甲基維斯阿米醇苷、蒙花苷對照品,依“2.2.1”項的方法制備相當于100%濃度水平的供試液,在2.1色譜條件下測定。對照品加入量及測定結果見表2。
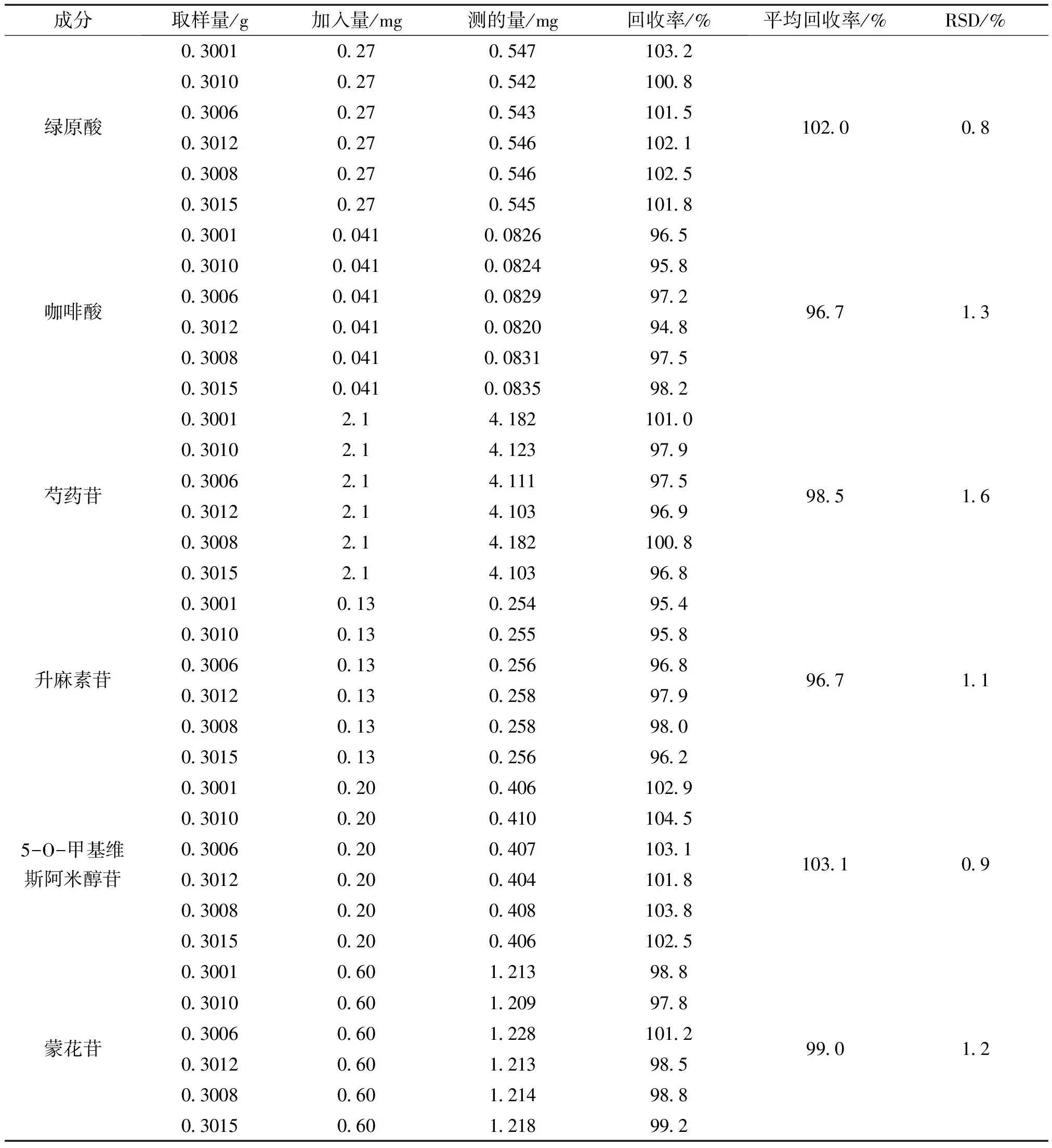
表2 加樣回收率實驗結果 (n=6)
2.4 樣品測定及結果分析 取防芷鼻炎片6批(R廠家:20220101,20230301;T廠家:220202,230101;B廠家:E22S003,E22S001),依“2.2.1”項下方法制成供試液,在“2.1”項條件下進行檢測,6批樣品中6種成分的含量測定結果折線圖如圖3所示。從圖中可看出:同一廠家不同批號的樣品的各成分的含量差異不顯著,而不同廠家生產的樣品各成分的含量都有差異,尤其是R廠家的樣品中芍藥苷、升麻素苷、5-O-甲基維斯阿米醇苷及蒙花苷的含量遠高于T和B廠家。
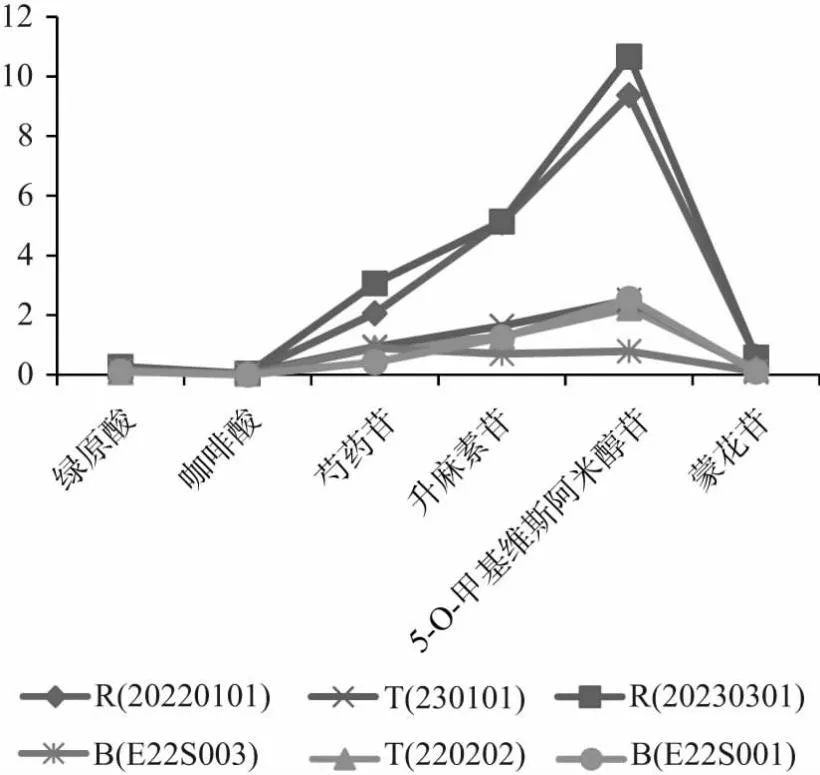
圖3 含量測定結果折線圖
3 討論
3.1 樣品溶液的制備 溶劑的選擇上,分別考察了70%乙醇溶液、100%甲醇溶液、甲酸-50%甲醇(1∶20)混合溶液和甲酸-70%甲醇(1∶20)混合溶液,70%乙醇溶液和100%甲醇溶液對綠原酸、咖啡酸的提取效率低,甲酸-50%甲醇(1∶20)混合溶液對各成分的提取效率明顯優于甲酸-70%甲醇(1∶20)混合溶液。提取方式上,比較了超聲和加熱回流兩種方法,與超聲提取的樣品比較,加熱回流提取的樣品中,蒙花苷的含量顯著提高,其他成分的含量略有提升。這可能與蒙花苷溶解性能有關,溫度較高的條件下,其在甲醇中的溶解度升高;基于上述原因,選取甲酸-70%甲醇(1∶20)混合溶液為溶劑,采用加熱回流的提取方式。
3.2 流動相及洗脫方式的選擇 流動相的選擇上,依綠原酸、咖啡酸等6種組分的化學性質,選取甲醇-水、甲醇-0.5%醋酸溶液和甲醇-0.1%磷酸溶液為流動相考察對象,發現:以甲醇-水為流動相時,樣品中綠原酸和咖啡酸峰面積小、峰形不佳;以甲醇-0.1%磷酸溶液為流動相時,芍藥苷和升麻素苷的峰形對稱性欠佳;而以甲醇-0.5%醋酸溶液洗脫,6個組分色譜峰的峰形對稱,分離度佳,且峰面積均較大;洗脫方式上,因各組分的極性相差較大,等度洗脫需要耗費更多的溶劑和時間,故選擇梯度洗脫。綜上所述,本實驗選取甲醇-0.5%醋酸溶液為流動相,進行梯度洗脫。
3.3 檢測波長的選擇 通過對防芷鼻炎片供試品溶液進行全波長掃描分析,確定了干擾小、強吸收的波長,分別為323 nm(綠原酸、咖啡酸)、230 nm(芍藥苷)、254 nm(升麻素苷、5-O-甲基維斯阿米醇苷)、330 nm(蒙花苷);查看樣品溶液的離線3D(時間、波長、峰面積)視圖,發現在21 min、30 min、60 min切換波長,所得的色譜圖基線平穩、色譜峰分離度高。
4 結論
使用HPLC梯度洗脫-切換波長法對3個廠家6個批號的防芷鼻炎片進行測定,結果表明,防芷鼻炎片中的6個指標性成分均能在同一個色譜系統中同時出峰,且結果穩定、分離效能高。因此,HPLC梯度洗脫-切換波長法可作為防芷鼻炎片6種成分的含量測定方法。此方法的建立,有利于更全面控制防芷鼻炎片的質量,但仍存在不足。該方法所能測定的指標成分并未覆蓋全方,且實驗的樣本量也較少。因此,今后可結合指紋圖譜和藥理實驗確定更多能反應該藥藥效的質量標志物,測定覆蓋全方中藥材的指標性成分含量,探索其成分之間的相關性,建立更高效更全面的含量測定方法。
