Pool-ARMS:一種高效檢測混合遺傳樣本中特定單核苷酸變異的方法
2024-01-20 01:52:22曹麗淼譚瑗瑗于清濤舒慶堯
核農學報 2024年2期
曹麗淼 譚瑗瑗 汪 慶 錢 秋 于清濤 舒慶堯,5,*
(1浙江大學現代種業研究所/水稻生物育種全國重點實驗室,浙江 杭州 310058;2浙江大學新農村發展研究院,浙江 杭州 310058;3無錫哈勃生物種業技術研究院有限公司,江蘇 無錫 214100;4哈爾濱市農業科學院,黑龍江 哈爾濱 150029;5浙江省作物種質資源重點實驗室,浙江 杭州 310058)
單核苷酸變異(single nucleotide variation,SNV),又稱單核苷酸多態性(single nucleotide polymorphism,SNP),廣泛存在于動植物基因組中,影響RNA 轉錄、剪切和蛋白質翻譯及其功能。在水稻中,Wx基因第一內含子剪切供體位點SNV(G→T)使粳稻稻米直鏈淀粉含量降低[1-2];一個編碼RNA 酶基因TMS5第71 位的一個SNV(C→A)產生了溫敏雄性不育特性[3]。迄今,已有多種SNV分析方法在作物育種中應用。如將SNV 轉化為基于酶切位點差異的酶切擴增多態性序列(cleaved amplified polymorphic sequence,CAPS)或衍生酶切擴增多態性序列(derived cleaved amplified polymorphic sequence,dCAPS)標記[4-5],基于DNA雙鏈熔解特性差異的高分辨熔解曲線(high-resolution melting analysis,HRM)技術[6],以及利用DNA聚合酶延伸對引物3'端或附近與模板的錯配高度敏感的特性建立的擴增阻滯突變系統PCR(amplification refractory mutation system PCR,ARMS-PCR)等[7]。其中,利用ARMS-PCR可以準確快速地對SNV進行分型,目前已成功應用于如水稻香味、光合效率相關基因[8-9]。但是,該方法只用于個體的基因分型,不能用于混合群體的突變檢測。
通過誘變或堿基編輯創制特異SNV 位點是培育優異種質資源最直接有效的手段。由于誘發突變的頻率極低,僅有7.5×10-6~9.8×10-6[10],美國科學家開發了一類混合樣本中檢測突變的技術,即定向誘導基因組局部突變技術(targeting induced local lesions in genomes,TILLING)[11],并衍生了如iTILLING(individualized TILLING)[12]、Seq-TILLING[13]、HRM-TILLING[14]等檢測不同類型突變的技術體系。這些技術除基于二代測序(next generation sequencing,NGS)技術的Seq-TILLING 外,植株混合檢測的倍數都很小(一般為1∶4)且檢測的是各類隨機突變,無法檢測特定SNV。
在基因組編輯研究中,已開發基于PCR 片段分型和測序的多種突變檢測技術[15]。但是,這些方法也無法對特定SNV 進行檢測[16]。最新開發的引導編輯技術(prime editing,PE)可以精確地實現所有核苷酸之間的替換,但靶點的唯一性使其在特殊位點的編輯效率仍然很低,從而嚴重限制了該技術在植物育種中的應用[17-19]。
為了建立可以高效檢出誘變和堿基編輯群體中攜帶目標SNV個體的技術體系,大幅降低兩類群體中目標個體的檢出成本,根據ARMS-PCR的基本原理,本研究設計一種適用于混合遺傳樣本中檢測攜帶特定SNV個體的方法,簡稱Pool-ARMS。以水稻兩個由單堿基變異引起的長鏈非編碼RNA基因(long non-coding RNA,lncRNA)PMS1和PMS3為例開展實證性研究[20-22],即PMS1的一個G→T(pms1)和PMS3的一個C→G(pms3)使水稻獲得了PGMS 特性,以期證明該策略的應用價值。
1 材料與方法
1.1 常規材料和PGMS水稻
本研究試驗材料使用粳稻包臺型細胞質雄性不育(BT-cytoplasmic male sterility,BT-CMS)保持系浙粳7B[23]的黃葉突變體浙粳7BY,浙江大學水稻生物育種全國重點實驗室(本實驗室)采用花藥培養育成的粳型溫敏不育系TS949,野敗型秈稻細胞質雄性不育系保持系龍特浦B的翠綠葉突變系翠玉B(CYB)[24],聚合抗稻瘟基因Pi1和Pigm的秈稻9311的近等基因系9311pg[25],以及粳型光周期敏感性雄性不育(photoperiod-sensitive genic male sterility,PGMS)系江79S[26]。
1.2 PGMS水稻的堿基編輯
為通過堿基編輯培育PGMS 水稻,本研究采用高效代理引導基因編輯器編輯目標SNV,在pegFinder 網站(http://pegfinder.sidichenlab.org)根據pms1和pms3的序列設計pegRNA 序列[27]。每一個表達片段都有2 個sgRNA(一個nicking sgRNA 用于切割未編輯的鏈以提高編輯效率,另一個作為pegRNA的一部分)、一個逆轉錄序列(reverse transcription,RT)和一個引物結合位點(primer binding site,PBS),使用多順反子tRNA的策略以提高多重編輯能力和效率,從而產生pegRNA和nicking sgRNA 的串聯序列[28]。用于這兩個基因引導編輯的pegRNA 和nicking sgRNA 序列具體信息見表1。分別合成了382和377 bp的pegRNA-tRNA-sgRNA-tRNA串聯序列(重復序列tRNA為77 bp,gRNA scaffold為76 bp)。為實現雙靶基因的引導編輯,將兩者合成為一個759 bp的表達片段。最后,將目的表達片段導入基于潮霉素和雙草醚抗性基因的雙代理引導基因編輯器PE3-DS載體[19]。具體方法為:Hind Ⅲ酶切PE3-DS 載體并回收,用In-fusion 無縫克隆方法將表達盒與PE3-DS 線性化載體連接,轉化大腸桿菌DH5α,菌落PCR 篩選陽性克隆。重組質粒經測序和序列比對,若正確則用于后期轉化。
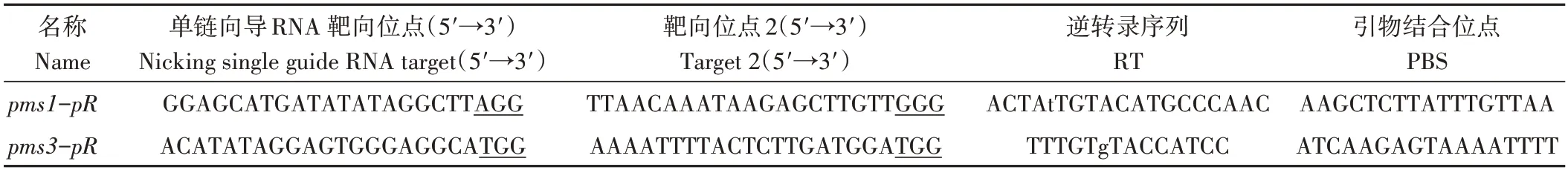
表1 用于PGMS基因PMS1和PMS3引導編輯的pegRNA和nicking sgRNA序列Table 1 pegRNA and nicking sgRNA sequences for prime editing of PGMS genes PMS1 and PMS3
從成熟胚誘導水稻愈傷組織,并用活化的攜帶堿基編輯重組質粒的農桿菌侵染生長狀態良好的亮黃色致密胚性愈傷組織[29]。農桿菌浸染的愈傷組織在培養基上暗培養3 d后,用無菌水和含頭孢的無菌水依次進行浸泡沖洗,瀝干后將轉到含500 mg·L-1頭孢和50 mg·L-1潮霉素的第一輪選擇培養基上,26 ℃、16 h 光照/8 h 黑暗條件下培養2 周。因為PE3-DS 載體是一個基于潮霉素Y46*和基于OsALSS627I組合的雙代理系統,因此在抗性愈傷組織第二輪篩選培養基將潮霉素濃度提高至80 mg·L-1的同時加入0.65 μmol·L-1的雙草醚(bispyribacsodium,BS),置于26 ℃、16 h光照/8 h黑暗條件下培養。之后,2 周換一次培養基,直至長出抗性愈傷組織。將抗性愈傷組織進行誘導分化和生根,獲得堿基編輯的T0植株,取1~2 cm葉片用于后期檢測。
1.3 擴增阻滯PCR引物設計與程序優化
采用十六烷基三甲基溴化銨(cetyltrimethylammonium bromide,CTAB)法提取水稻基因組DNA[30]。根據ARMS-PCR原理,采用Primer Premier 5.0軟件設計一條與模板DNA 序列完全一致的引物,即用于擴增PMS1/pms1的pms1/R 和用于擴增PMS3/pms3的pms3/F,一條用于區別目標SNV與非目標SNV等位基因的帶錯配堿基的引物(圖1)。設計攜帶錯配的引物時,3'末端的最后一個核苷酸與目標SNV 一致,在3'末端倒數第二位或第三位引入一個錯配堿基,錯配堿基為除本身堿基外另三種堿基類型(圖1)。
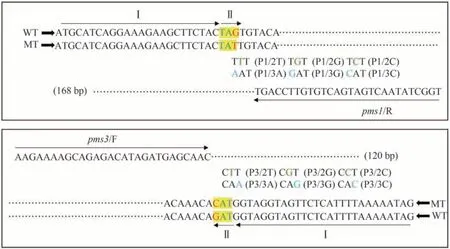
圖1 兩個水稻PGMS基因(PMS1和PMS3)攜帶SNV的片段及其ARMS-PCR引物設計示意圖Fig.1 Diagram of the fragments of two rice PGMS genes PMS1 and PMS3 and their respective SNVs and the primers designed for ARMS-PCR
為確定最佳引物和擴增條件,采用梯度PCR進行擴增。PCR 體系為20 μL,體系為:引物各0.8 μL,Green Taq Mix(P131,南京諾唯贊生物科技股份有限公司)10 μL,無菌水7.4 μL,模板1 μL(200 ng·μL-1)。擴增程序為:95 ℃預變性3 min;95 ℃變性15 s,退火15 s(溫度梯度為56~66 ℃,PMS1為62 ℃,PMS3為63 ℃),72 ℃延伸,共30個循環;72 ℃延伸5 min。PCR產物1 %瓊脂糖凝膠電泳,拍照保存。
經檢測擴增成功的PCR 產物送浙江有康生物科技有限公司進行Sanger 測序,使用DNAMAN 6.0 軟件分析每個目標位點的Sanger測序圖譜。
1.4 混池樣本篩選SNV檢測
為建立混合樣本SNV檢測體系,以浙粳7BY(PMS1和PMS3)和江79S(pms1和pms3)為材料,按三種方式進行DNA 混樣:1)DNA 混樣:先提取浙粳7BY 和江79S葉片的DNA,按1∶9、1∶19、1∶49、1∶99的比例混合成樣;2)葉片混樣:用1.5 mm 打孔器取單株葉片樣本,再按不同比例混合制成葉片樣本,提取DNA;3)芽鞘混樣:將種子浸種2 d、催芽2 d以獲得萌動的種子,取其胚芽鞘4 mm,再按不同比例混合制成芽鞘樣本,提取DNA。葉片混樣和芽鞘混樣兩種混合樣本中江79S∶浙粳7BY的比例設為1∶1、1∶9、1∶19、1∶24、1∶49。以1.3確定的最佳ARMS-PCR條件檢測目標SNV。
2 結果與分析
2.1 不同水稻材料的PMS1和PMS3基因型
為保證試驗材料的準確性,先對包含PMS1和PMS3基因SNV 位點的片段測序。結果證實,江79S的SNV-pms1和SNV-pms3分別為T和G,與已報道的PGMS材料一致;其余四份材料的SNV-pms1和SNV-pms3分別為G和C,均為野生型PMS1和PMS3(圖2)。

圖2 不同水稻材料PGMS基因PMS1和PMS3片段序列分析Fig.2 Sequence analysis of PGMS genes PMS1 and PMS3 in different rice accessions
2.2 PGMS等位基因特異性擴增
為建立PGMS 等位基因特異性擴增體系,以浙粳7BY 和江79S 兩種材料,分析了12 對PCR 引物特異性擴增光敏SNV 的效果。結果發現,在56~64 ℃溫度范圍內,用倒數第三位堿基錯配的引物擴增可以明顯區分PMS1和pms1;在61~63 ℃溫度范圍內,用倒數第三位堿基錯配的引物擴增可以明顯區分PMS3和pms3。在這兩個PGMS基因位點上,用3'末端倒數第二位堿基的錯配的引物區分效果均差于倒數第三位堿基錯配的引物。
為確定最佳退火溫度以及錯配堿基的類型,對用倒數第三位堿基錯配的引物區分SNV 的效果做了進一步研究。發現倒數第三位堿基錯配為G的引物區分PMS1/pms1和PMS3/pms3的效果最好,最佳退火溫度分別為62 和63 ℃。因此,建立了特異性擴增PGMS 等位基因pms1和pms3的引物和PCR參數(表2)。

表2 PCR擴增條件的參數Table 2 Optimized PCR amplification conditions
采用以上引物和擴增條件,多次對4份非PGMS材料進行擴增,結果均無擴增條帶(圖3),說明可以特異且高效檢測PGMS基因。

圖3 水稻PGMS基因pms1和pms3等位基因型檢測Fig.3 Genotyping of two PGMS genes pms1 and pms3 in rice
2.3 混合樣本SNV檢測
2.3.1 葉片組織DNA樣本 葉片是提取DNA最常用的材料。比較先提取DNA 再混合成DNA 池和取等量葉片組織混合后直接提取DNA 兩種方法的檢測效果。采用先提取再根據DNA 濃度調整后按不同比例混合形成混樣DNA模板,發現以1∶99混合時仍可擴增出特異性條帶(圖4)。在采用打孔器從葉片中均勻取樣、混合,提取的DNA 用于PCR 模板時,無論是pms1還是pms3,檢出水平降為1∶49(圖5)。上述方法均經過多輪、多次試驗,結果均能很好重現。

圖4 葉片提取DNA按不同比例混合成樣本的突變檢測電泳圖Fig.4 Mutation detection of mixed DNA from leaves samples in different proportions using agarose electrophoresis

圖5 不同比例葉片混合樣本的突變靈敏度檢測電泳圖Fig.5 Mutation detection of mixed leaf samples in different proportions using agarose electrophoresis
2.3.2 萌動種子芽鞘組織混合DNA 樣本 由于芽鞘組織量很少,保證取完胚芽鞘后的種子仍能正常生長,本研究采用先混合芽鞘組織的樣本、再提DNA 的方法。多次重復試驗結果表明,pms1和pms3位點的檢出效率為1∶24的比例(圖6)。

圖6 不同比例胚芽鞘混合樣本的突變檢測電泳圖Fig.6 Mutation detection of mixed coleoptile samples in different proportions using agarose electrophoresis
2.4 堿基編輯材料的pms1和pms3 SNV檢測
本試驗共獲得pms1和pms3雙靶點基因編輯載體轉基因T0植株63株,其中CYB轉化苗15株,浙粳7BY轉化苗48株。采用打孔器葉片取樣后提取單株的DNA,利用本研究建立的特異性PCR體系,擴增并檢測目標條帶。結果顯示,試驗組均沒有擴增出特異性條帶(圖7)。為驗證Pool-ARMS 結果的準確性,對63株植株單獨提取DNA 并采用常規方法擴增目標片段,進行Sanger 測序分析。結果顯示(圖8),這些植株的PGMS基因與親本一致,均為正常可育基因型,與Pool-ARMS 法確定的結果一致。

圖7 兩個PGMS基因堿基編輯材料的Pool-ARMS檢測電泳圖Fig.7 Pool-ARMS detection for the mutation of two PGMS genes in base editing plants
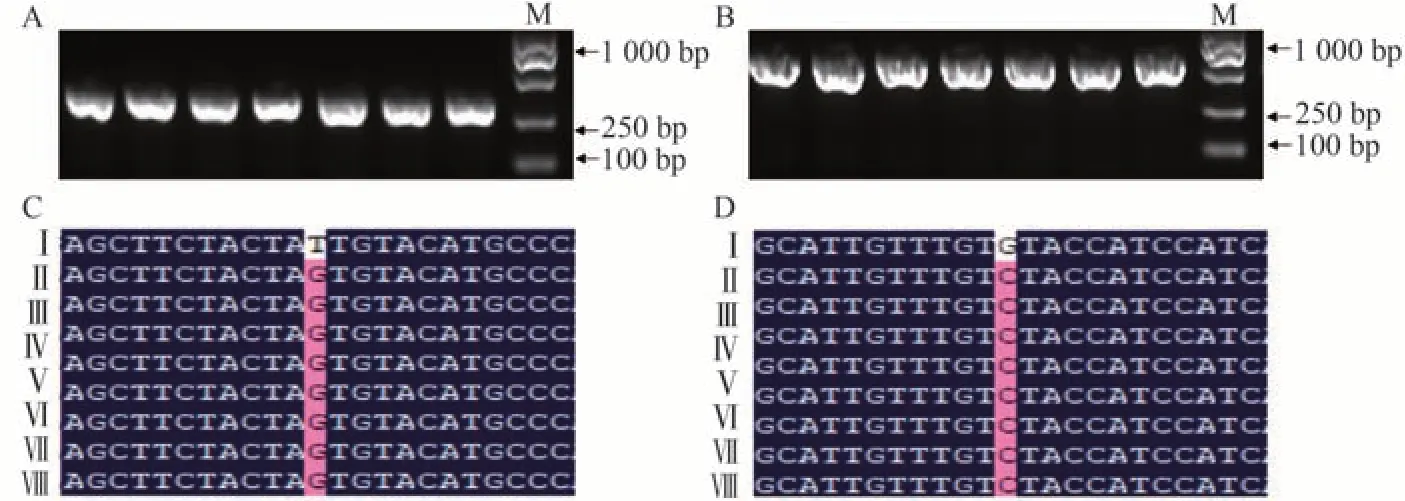
圖8 兩個PGMS基因PMS1(A)和PMS3(B)堿基編輯水稻T0植株的PCR片段和序列比對圖Fig.8 PCR amplification and sequence alignment of two PGMS genes PMS1(A)and PMS3(B)in T0 rice plants transformed with base-editing vectors
3 討論
ARMS-PCR 作為一種操作簡便、準確性較高且低成本的SNV 分型技術,可以準確地將單堿基突變的樣本進行區分,實現快速、簡單、低成本和高通量的基因分型[31],具有廣闊的應用前景和發展潛力。在特異性SNV 篩查中,只需確定群體中是否含有特定SNV 的個體,無需對特異和非特異SNV基因型進行分型,因此目標是建立特異性擴增目標SNV的體系。基于這一需求,本研究通過針對目標SNV 設計一條特異擴增引物,即在該引物的3'末端的倒數第三位堿基處增加一個錯配,結合適宜的退火溫度,實現了對群體中特異SNV的靈敏檢測。
目前,多數基因檢測技術都是用于個體等位基因分型,尚缺少可以用于群體中特異SNV 篩查的廉價技術體系。在醫藥和環境科學中,數字PCR(digital PCR,dPCR)可以對群體中稀有突變等位基因進行定量分析。但是,dPCR儀器昂貴,所需試劑耗材成本高,需要事先優化流程并研制出相應的探針,該方法在植物育種中的應用前景尚有待進一步研究。深度測序技術理論上也可以用于混池樣本稀有突變的檢測,但在實際應用中依賴具有生物信息分析技能的專業人員,而且該方法檢出的假陽性比例較高,對篩查出的混樣需要通過單個樣本的進一步分析后確定[32]。因此,建立基于普通PCR 程序、操作簡便快速、對樣品的純度要求較低的混樣SNV 檢測技術體系,具有廣泛的應用前景。本研究將ARMS-PCR 的原理應用于群體SNV 的檢測,建立了Pool-ARMS 體系,該方法可以大幅提高檢測效率并縮小成本,為篩選特定SNV 建立了一種高效、低成本的方法,在突變群體和堿基編輯植株特定SNV篩查中具有良好的應用前景。
本研究針對實際應用場景確定了利用葉片和芽鞘組織篩選特定SNV 個體的技術體系。對水稻堿基編輯植株這樣的群體,可以通過等量混合葉片樣本提取DNA,開展Pool-AMRS分析,在50株T0植株的混樣中檢出目標SNV。對水稻誘變群體,特定SNV的比例極低,需要對高達10萬個M2個體開展檢測[10]。本研究建立的利用芽鞘組織的技術體系,可以實現當天取樣,當天或次日出結果,從而減少田間種植、取樣這一費時費力、易錯的環節,大幅簡化突變體篩選流程。按照1∶24 的檢出能力,10 萬粒種子僅需建立約4 000 個混樣,進行約400 次PCR 和相應的瓊脂糖電泳檢測,將工作成本變得可控。
本研究建立的pms1和pms3特異SNV 檢測體系主要用于混池群體的檢測,同時也可以在其他育種方式中應用。例如,在水稻的花培育種中,加倍單倍體植株為純合野生型或純合突變型。因此,利用本研究的篩選方法,如果一個植株同時檢測到pms1-SNV 和pms3-SNV,可以推定該材料即為PGMS水稻。
4 結論
本研究以堿基編輯和誘發突變的群體為目標應用場景,以控制光周期敏感性雄性不育(PGMS)水稻SNV為例,優化PCR引物和反應程序,建立了一種通過混池篩查高效檢出目標SNV 的方法Pool-ARMS。利用葉片和芽鞘組織混樣提取DNA 檢測突變的體系,將突變檢出水平分別確定為1∶49和1∶24,實現經濟、高效、簡便的目標,可以運用到水稻等農作物育種特別是利用誘發突變和堿基編輯創制種質資源中,實現對目標變異的定向篩選。
猜你喜歡
青少年科技博覽(中學版)(2022年6期)2022-12-27 19:44:27
今日農業(2021年21期)2021-11-26 05:07:00
軍事文摘(2021年22期)2021-11-26 00:43:51
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
今日農業(2021年14期)2021-10-14 08:35:40
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
文苑(2020年6期)2020-06-22 08:41:52
