單細胞轉錄組學技術及其在寄生蟲研究中的應用進展
2024-11-08 00:00:00周蜓蜓李立吳燕濤逯文穎付寶權殷宏賈萬忠閆鴻斌
畜牧獸醫學報 2024年10期
關鍵詞:數據分析
摘 要: 單細胞轉錄組代表了某一時刻單個細胞內所有mRNA總表達量,其表達量反映該細胞的總體特征。通過單細胞轉錄組測序(single cell RNA sequencing, scRNA-seq)技術對單個細胞的全部RNA進行逆轉錄、擴增和測序,測序結果中包含的大量信息有可能重塑對發育生物學、基因調控以及健康和疾病中細胞異質性的理解。隨著科學技術的不斷發展,測序技術也在不斷進步,scRNA-seq全方位、高通量以及高分辨率的特點可構建更細致、全面和精準的單細胞圖譜。本文綜述了常用scRNA-seq方法及其數據分析主要流程和單細胞轉錄組學技術在寄生蟲學與寄生蟲病領域研究中的應用,以期為該領域相關研究提供參考資料。
關鍵詞: 單細胞測序;轉錄組;數據分析;寄生蟲病
中圖分類號:S855.9;Q522.6
文獻標志碼:A
文章編號:0366-6964(2024)10-4290-12
收稿日期:2023-12-25
基金項目:國家重點研發計劃(2022YFD1800200);甘肅省科技重大專項(23ZDNA007;22ZD6NA001);甘肅省在站博士后專項(23JRRA558);國家肉牛牦牛產業技術體系項目(CARS-37);
蘭州大學中央高校基本科研業務費專項資金(lzujbky-2024-ydyl02);動物疫病防控全國重點實驗室重大成果培育項目(SKLADCP2023HP05);
中國農業科學院創新工程(CAASTIP-2024-5;CAAS-ASTIP-2021-LVRI)
作者簡介:周蜓蜓(1999-),女,重慶云陽人,碩士生,主要從事棘球蚴病發育與致病機制研究,E-mail:1305403647@qq.com
*通信作者:閆鴻斌,主要從事棘球蚴病學研究,E-mail:yanhongbin@caas.cn;賈萬忠,主要從事動物絳蟲(蚴)病研究,E-mail:jiawanzhong@caas.cn
Progress on Single-cell Transcriptomics Technology and Its Applications in Research
on Parasites
ZHOU" Tingting1, LI" Li1, WU" Yantao1, LU" Wenying1, FU" Baoquan1, 2, YIN" Hong1, 2, JIA" Wanzhong
1, 2*, YAN" Hongbin1*
(1.State Key Laboratory for Animal Disease Control and Prevention/College of Veterinary Medicine, Lanzhou University/National Para-reference Laboratory for Animal Echinococcosis/Gansu Province Research Center for Basic Disciplines of Pathogen Biology/Key Laboratory of Veterinary Parasitology of Gansu Province/Key Laboratory of Veterinary Etiological Biology and Key Laboratory of Ruminant Disease Prevention and Control (West), Ministry of Agricultural and Rural Affairs/Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou 730000, China; 2.Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Disease, Yangzhou 225009, China)
Abstract: The single cell transcriptome represents the total expression of all mRNA in a single cell at a certain time, and its expression level reflects the overall characteristics of the cell. The entire RNA of a single cell is reversely transcribed, amplified and sequenced by single cell RNA sequencing (scRNA-seq). The extensive information contained in the sequencing results has the potential to reshape the understanding of developmental biology, gene regulation and cellular heterogeneity in health and disease. With the continuous development of science and technology, sequencing technologies are also constantly improving. The omni-directional, high-throughput and high-resolution characteristics of scRNA-seq can build a more detailed, comprehensive and accurate single cell map. This paper summarizes the commonly used scRNA-seq methods, the main processes of data analysis, and the application of single cell transcriptome technology in the field of parasitology and parasitic diseases, in order to provide reference materials for related research in this field.
Key words: scRNA-seq; transcriptome; data analysis; parasitic diseases
*Corresponding authors:" YAN Hongbin, E-mail: yanhongbin@caas.cn; JIA Wanzhong, E-mail: jiawanzhong@caas.cn
近40年來,用于分析RNA的技術逐步改進并持續推動著生命科學的進步。在20世紀80年代,研究人員已經能夠通過Northern技術(RNA印跡)進行核酸定量,聚合酶鏈式反應(polymerase chain reaction, PCR)及隨后的逆轉錄聚合酶鏈式反應(reverse transcription-PCR, RT-PCR)的發現,進一步推動了相關學科的發展[1],同時,對mRNA的分析提供了關于細胞和組織特異性基因表達特征的詳細信息[2]。1992年,Eberwine等[3]首次提出在單細胞水平上對整個轉錄組進行測序,通過擴增特定單細胞的RNA,構建了大鼠海馬中單個活細胞的基因表達譜,并發現了一些未被描述的mRNA。2009年,Tang等[4]基于下一代測序技術進行了首次單細胞轉錄組測序分析,發現在單細胞分辨率下獲得基因表達譜是可行的,并描述了早期發育階段細胞的特征。這項研究為單細胞RNA測序的概念和技術方法帶來了巨大突破,實現了細胞測序數量的擴大,使高通量RNA測序更兼容,意味著可以從整個組織和單個細胞中識別和量化RNA轉錄本。隨著測序技術的進步,單細胞轉錄組測序(single cell RNA sequencing, scRNA-seq)技術也在不斷發展,在單項研究中已經能夠達到百萬細胞水平的轉錄組測序[5],這對發現和辨別稀有細胞類型具有重要意義,使人們能夠在轉錄組水平上對細胞進行分類、鑒定、表征和區分,也能夠在細胞水平上揭示各類疾病的發展過程。
寄生蟲病是全球性公共衛生問題之一,不僅威脅全球畜牧業健康發展,影響動物生長發育,降低養殖效益,而且有些寄生蟲還可以感染人類,嚴重威脅人類健康[6],探究寄生蟲的生長發育過程及其與宿主的相互作用機制,可以為寄生蟲病防控提供重要的理論依據。scRNA-seq具有無偏倚、高通量和高分辨率的特點,能夠為寄生蟲學相關研究提供更加細致嚴謹的研究技術。通過scRNA-seq分析單個細胞的基因表達差異,可以在單細胞水平上探究寄生蟲與宿主細胞之間的相互作用關系,揭示不同類型細胞對寄生蟲生命周期的影響以及寄生蟲引起的宿主免疫應答,還可以檢測和鑒定新的寄生蟲物種[7-8],進一步解析寄生蟲的細胞結構,并從基因層面探究寄生蟲的發育機制[9]。本文主要綜述scRNA-seq技術的主要原理和數據分析方法,并重點介紹scRNA-seq技術在寄生蟲研究領域中的應用進展。
1 單細胞轉錄組測序技術
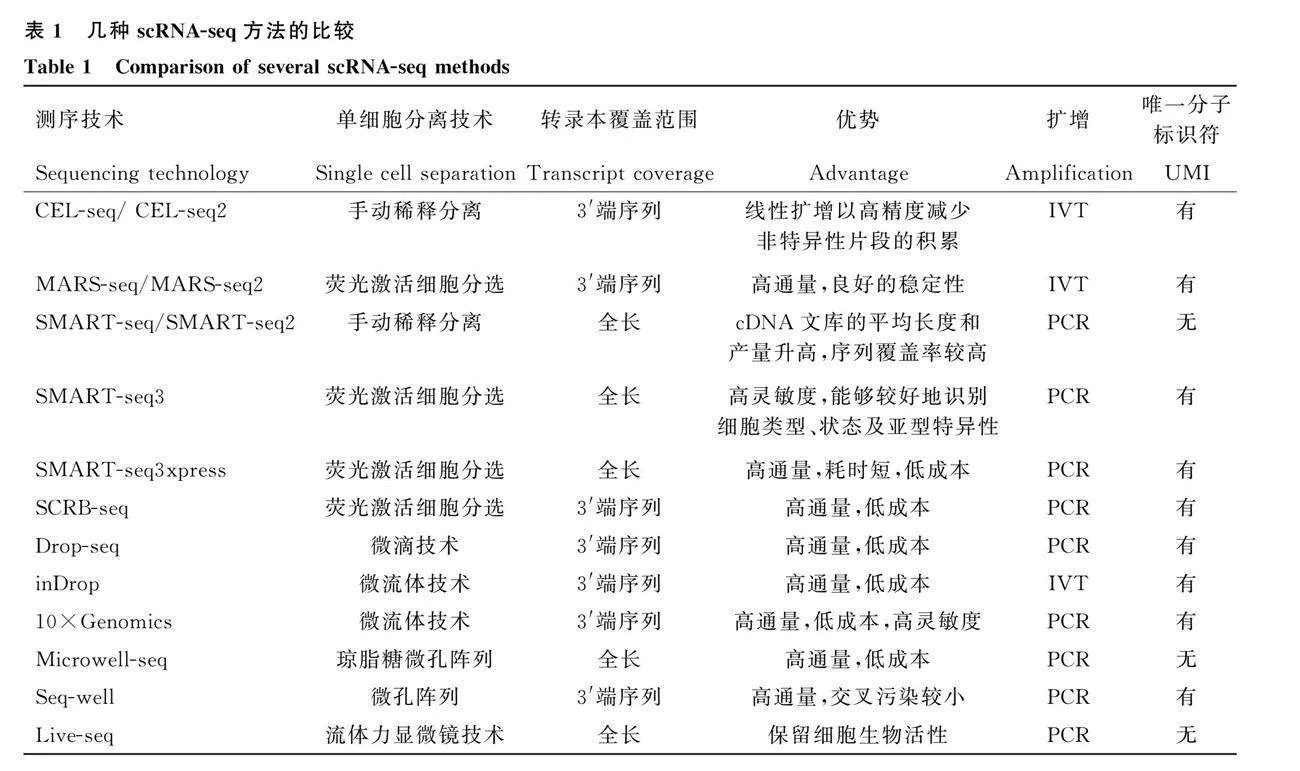
scRNA-seq的試驗流程通常需要將目的器官或組織分離為單個細胞,在此過程中需要最大限度地提高細胞數量和細胞質量,同時盡可能減少消化時間以降低細胞的死亡率。目前,已經發展成熟的單細胞分離技術包括稀釋法、顯微操作法、激光捕獲顯微切割法、熒光激活細胞分選(fluorescence activated cell sorting, FACS)和微流控技術等,其中稀釋法和顯微操作法能夠較好地保留細胞的完整性,但其具有通量低和耗時長的特點,因此主要針對于細胞數量較少的樣本組織。FACS和微流控技術是目前最常用的單細胞捕獲方法,二者的特點是高通量、自動化和低成本地進行單細胞分選,其中FACS還具有高特異性的特點[10]。隨后對捕獲的單細胞進行RNA的逆轉錄,并對cDNA實施擴增從而進行文庫制備,再通過下一代測序生成讀數,這些讀數與參考基因組對齊,進行處理及質量控制后對數據進行分析[11]。單個細胞不同的分離技術會導致讀取的mRNA濃度與實際濃度具有差異,同時由于擴增技術的不同,測序數據的質量也會產生差異[12],因此,在實際工作中時,應當充分考慮需要捕獲和測序的細胞數量,并綜合試驗目的來設計和執行scRNA-seq技術,當前常用的scRNA-seq技術對比見表1[13-27]。對于部分技術不同的擴增方式、測序原理的差異及其特點總結如下。
1.1 基于非液滴技術的單細胞測序
線性擴增單細胞測序(cell expression by linear amplification and sequencing, CEL-seq)是通過多重線性擴增進行scRNA-seq的技術,Hashimshony等[13]為了應對RNA起始量較小這一挑戰,通過條形碼和匯集樣本,使用體外轉錄(in vitro transcription, IVT)線性擴增mRNA,從而開發了CEL-seq。該方法將分離的單個細胞轉移至低滲裂解液中進行裂解,獲得含有mRNA的裂解緩沖液,再通過添加含有唯一條形碼、Illumina 5′接頭和T7啟動子的oligo dT引物、逆轉錄酶和脫氧核糖核苷三磷酸等,對mRNA進行逆轉錄,獲得cDNA第一條鏈。隨后對cDNA進行轉錄獲得用于IVT反應的模板材料,由此擴增的RNA能夠進行雙向的RNA文庫構建。具體步驟為將RNA片段化成適合測序的大小,通過連接添加Illumina 3′接頭,將RNA反轉錄為cDNA,最終從文庫中篩選出同時包含Illumina 3′接頭、Illumina 5′接頭和條形碼的大多數片段。得到的文庫經過配對測序,其中第一次讀取恢復條形碼,第二次識別mRNA轉錄本,達到識別單細胞RNA的目的。相較于PCR擴增方法,IVT線性擴增的主要優勢在于其重復性高,錯誤率低,但其生成文庫所花費時間更長。2016年,CEL-seq經過優化發展為CEL-seq 2[14],將唯一分子標識符(unique molecular identifiers, UMI)加入到CEL-seq引物中,使每個反轉錄的mRNA能夠精確計數一次,并將CEL-seq引物從92個核苷酸縮短到82個核苷酸用以提高逆轉錄(reverse transcription, RT)反應的效率,此外還在RT步驟中直接插入Illumina適配器作為5′端,隨機連接到六聚體,從而省去了以往的連接步驟,提高靈敏度的同時進一步減少了文庫準備的實際操作時間和成本。
大規模平行單細胞測序(massively parallel single-cell RNA sequencing, MARS-seq)也是一種常用的單細胞轉錄組建庫和測序技術,與CEL-seq通過稀釋法獲得單細胞不同的是,MARS-seq采用FACS將單個細胞分選至384孔板,隨后的技術原理與CEL-seq相似,通過T7啟動子連在oligo dT引物上,cDNA合成后再啟動IVT生成帶有條形碼和UMI的擴增RNA[15]。為了提高實驗的穩定性和檢測率,Keren-shaul等[16]發展的MARS-seq 2對MARS-seq單細胞處理的各個步驟進行了優化改進,包括修改RT引物的組成,降低RT引物的濃度,減少RT和裂解緩沖液的體積等,這種文庫制備方法,使生產成本顯著降低,且對于任意數量的細胞測序,單個細胞的成本都不會受到影響。因此,與其他scRNA-seq技術相比,在測定細胞數量較少的樣本組織時,MARS-seq2優勢在于具有較低的測序成本。此外,由于MARS-seq和CEL-seq在3′端引入標記,無法測定全長mRNA序列,所以不可用于可變剪切的分析,更適用于對基因表達進行定量[28]。
1.2 基于液滴技術的單細胞測序技術
在制備大量cDNA文庫過程中,實驗通量往往是scRNA-seq實驗設計中需要考慮的問題,少量細胞的RNA-seq數據通常不足以全面地反映生物樣品的狀態,因此大部分scRNA-seq將面臨數以萬計的細胞數量,而液滴微流體能夠以較高的頻率實現單個細胞的快速分隔和封裝,所產生的每個液滴僅有納升體積以適應單細胞反應,其液滴數量可達到數百萬個[29]。基于液滴策略的scRNA-seq系統能夠大幅度提高反應通量、降低成本,Macosko等[22]由此設計發展了單細胞高通量液滴測序(Drop-seq)。Drop-seq用納升大小的液滴包裹單個細胞和具有分子條形碼的微珠,細胞在液滴中裂解后釋放的mRNAs與微珠中的分子條形碼進行雜交,將液滴破碎后收集mRNAs雜交物,并對其進行逆轉錄從而形成mRNA逆轉錄產物,隨后對該逆轉錄產物進行反向轉錄、擴增和測序,最終使用逆轉錄產物條形碼來推斷每個轉錄本的細胞來源。
微滴高通量單細胞測序(inDrop)是另一種常用的基于液滴的高通量scRNA-seq系統[23],其構建了一種條形碼水凝膠微球(barcoded hydrogel microspheres, BHM),每個BHM中包含約109個共價連接的光釋放引物,并且編碼147 456個條形碼中的一個。再使用具有四個入口的微流控裝置,將裂解緩沖液、BHM、細胞和油進行混合,把細胞包裹成小滴,細胞封裝后在小滴中裂解,同時在合成cDNA的過程中被條形碼編碼,隨后將液滴打破使所有細胞材料混合在一起,并使用CEL-seq的方法對cDNA文庫進行測序[13]。相較于Drop-seq,inDrop采用BHM,可以將引物固定在微珠里面,與細胞混合后,通過紫外線曝光來釋放引物,而Drop-seq的引物只能固定在微珠表面,不能從微珠中釋放,導致其mRNA捕獲效率較低。盡管如此,面對成千上萬的細胞數量時,Drop-seq和inDrop仍然具有高通量和低成本的優勢。
1.3 新型單細胞測序技術
目前,大多數scRNA-seq技術都需要將細胞裂解再進行測序,這阻礙了對相同細胞分子特征和生物學功能的分析,為了克服這一難題,Chen等[25]創建了Live-seq方法,可在保持細胞存活的同時對其細胞質中的RNA進行提取,使細胞能夠用于后續研究。該方法的關鍵技術是流體力顯微鏡技術[30],該技術能夠在不裂解細胞的情況下,對細胞中部分細胞質進行采集。而Live-seq通過減少提取時間、降低溫度以及預加載帶有采樣緩沖液的流體力顯微鏡探針,達到立即將提取的細胞質液與RNase抑制劑混合的目的,并且能夠最大限度地再將提取液釋放到含有與下游RNA-seq兼容的緩沖液微液滴中,從而實施基于圖像的細胞跟蹤以用于順序提取。RNA轉錄本5′端的切換機制測序是測量少量RNA最靈敏的RNA-seq方法之一[31],Chen的團隊將該方法進行優化后,最大限度地從提取的mRNA中產生cDNA[32],最終僅利用1 pg的RNA完成建庫。總之,Live-seq使scRNA-seq不再是細胞研究的終點,而是成為揭示細胞分子生物學特征的有力證據,為單細胞轉錄組學技術提供了新的工具。
2 數據分析
單細胞轉錄組測序結束后通常會生成FASTQ格式的原始數據,在數據預處理過程中,細胞條形碼和UMI首先作為標簽附加到每個cDNA讀數的前端中,該步驟常用的工具包括CellRanger[33]和STARsolo[34]等,隨后cDNA需要通過映射工具將讀數映射到參考基因組或轉錄組[35]。Engstrm等[36]系統性地評估了26種RNA-seq序列比對方案,發現大多數方案在低于5個讀取覆蓋的連接處靈敏度降低,這反映了對齊算法依賴于交叉點覆蓋率來提高精度,然而無論連接覆蓋率的高低,基于注釋的基因組短讀核苷酸比對計劃方案都實現了高靈敏度。Dobin等[34]開發的STAR軟件,在映射速度方面比其他比對工具高出50倍,同時比對靈敏度和精度均有所提高,在適度的12核服務器上,每小時能夠對人類基因組進行5.5億次2×76 bp的配對端讀取。由于讀數的映射率是反映測序數據質量的重要指標[37],因此在測序技術和試驗方案不斷發展的前提下,必須開發新的映射策略,同時比對工具也應該提供靈活的框架來應對新的比對挑戰。
預處理后每個樣本被匯編成基因/條形碼矩陣,隨后可以進一步過濾和分析,分析scRNA-seq數據的整個工作流程包括讀取數據、去批次效應、整合、質量控制、基因表達定量、歸一化、特征選擇、降維、細胞聚類、擬時序分析和差異基因分析等[38]。由于scRNA-seq的局限性,包括轉錄本覆蓋的偏差、低捕獲效率和測序覆蓋率低等,在數據捕獲過程中會從破碎、死亡或與多個細胞混合的細胞中生成一部分低質量數據,這些數據會阻礙下游的分析,并可能導致對數據的誤解[37],因此,scRNA-seq數據的質控對于識別和去除低質量細胞至關重要。隨著單細胞測序技術的突飛猛進,已經開發了許多統計方法來處理scRNA-seq分析的不同步驟,常用的工具包包括基于R的Seurat[39]和Monocle 3[40]、基于python的Scanpy[41]等,這些工具包基于不同的算法設置參數,在進行數據分析時,可以通過更改其中的參數設置達到適用的分析效果。隨著技術的進步,分析軟件也在不斷更新優化,在未來有望將單細胞轉錄組學和其他組學相結合,實現多組學數據的綜合分析。
3 單細胞轉錄組學技術在寄生蟲病防控技術研究中的應用
3.1 在寄生蟲發育生物學研究中的應用
寄生蟲通常被定義為寄生關系中受益的一方,與地球上其他生物相同,寄生蟲也具有生物多樣性[42]。在原蟲研究中,阿米巴滋養體作為單細胞原生動物,常規的批量轉錄組測序無法揭示單個生物體的潛在變化,Feng等[8]使用scRNA-seq研究了單個阿米巴原蟲滋養體的RNA轉錄圖譜,發現與中國倉鼠卵巢細胞體外共孵育后,滋養體差異表達基因水平明顯升高,認為滋養體在與中國倉鼠卵巢細胞體外共孵育時處于活躍的轉錄狀態,同時發現其上調基因與半胱氨酸代謝相關,并且滋養體在半胱氨酸缺乏的條件下吞噬作用和細胞毒性顯著減弱。該研究發現隱蔽的滋養體特異性致病性變化,提高了對阿米巴滋養體生物學特性和毒力變異機制的理解。
在線蟲研究方面,秀麗隱桿線蟲是研究胚胎發育、繁殖和衰老的常用模式生物,Tintori等[43]通過scRNA-seq繪制了秀麗隱桿線蟲由胚胎到16-細胞階段的細胞圖譜,并提供了交互式數據可視化工具。隨后,Lorenzo等[44]基于秀麗隱桿線蟲的scRNA-seq數據設計了迭代聚類分析,成功將神經細胞亞型與解剖學定義的神經元類別進行匹配,并確定了新的神經元特異性啟動子。此外,Taylor等[45]制作了秀麗隱桿線蟲整個神經系統中所有神經元類型的scRNA-seq圖譜,共70 296個神經元圖譜,包括所有118個典型神經元類別,發現每個神經元類別都是由神經肽編碼基因和神經肽受體的不同組合定義的,這表明每種類型的神經元在發送和接收信號方面發揮著不同的作用,這些研究聚焦于整個神經系統的結構和功能與神經元特異性基因表達的全面聯系,為解析神經元特異性功能提供了理論依據。利用scRNA-seq技術,能夠獲得寄生蟲單個細胞的靶序列,進而從物種多樣性、生殖發育多樣性和遺傳多樣性等方面了解寄生蟲的生長及發育機制。
在扁形動物渦蟲研究中,2018年Fincher等[46]利用Drop-seq生成了66 783個地中海圓頭渦蟲(Schmidtea mediterranea)細胞的數據,共聚類出44個細胞亞群,定義了以前未知的渦蟲細胞類型,并闡明了干細胞和分化細胞間的轉換狀態,還發現肌肉中具有能夠提供位置信息的表達基因。渦蟲的修復能力強,是源于蟲體內一種稱為neoblast(成體多能干細胞)的成體干細胞群。而neoblast也是成體渦蟲體內唯一具有增殖和分化潛能的干細胞,能夠拯救動物受致命輻射傷害的neoblast被命名為克隆性成體干細胞(clonogenic neoblast, cNeoblast),從而與那些無法拯救受傷動物的neoblast相區分。Zeng等[47]針對渦蟲neoblast,利用10×Genomics大規模scRNA-seq技術發現一個新亞群neoblast 2,該亞群高表達PIWI-1基因和蛋白,并以保守的細胞表面蛋白(跨膜四蛋白)編碼基因1(tetraspanin 1, tspan-1)為標志,tspan-1的RNA干擾既影響新生細胞的再集落,又影響新生細胞的動員。重要的是,tspan-1陽性細胞在亞致死劑量的照射下存活下來,并進行克隆性擴增,以重新繁殖整個動物,滿足cNeoblast的定義,這為詳細解析渦蟲體內多能性和全身再生的潛在分子以及細胞機制打開了大門。此外,Wurtzel等[48]將渦蟲咽后切開后在不同時間點分離創口,并利用scRNA-seq獲得渦蟲傷口組織的單細胞圖譜,共619個渦蟲細胞,確定了13種不同的細胞類型,揭示了渦蟲再生的時間模型:在普通創傷反應達到頂峰后的24 h內會出現專門的轉錄程序,如果受傷后不需要再生,則創傷反應就會下降,否則就會出現特定傷害反應的表達,這些反應涉及模式分子和neoblast相關的特異基因,針對正在再生的組織特性,損傷后約3 d,與功能性新組織相關的分化組織標志物開始表達。研究渦蟲的再生問題,還能夠為實現人類器官的再生提供數據支撐。scRNA-seq技術為繪制寄生蟲不同生命階段細胞圖譜提供技術支撐,為寄生蟲個體的研究提供強有力的方法,也豐富了人們對寄生蟲生物學多樣性的理解。
3.2 在寄生蟲病疫苗研發中的應用
世界衛生組織的數據顯示,全世界有超過3億人患有一種或多種寄生蟲病,并且寄生蟲感染每年導致數百萬人死亡和殘疾[49]。盡管抗寄生蟲藥物能夠有效治療寄生蟲感染,但大部分藥物并不能完全殺死寄生蟲[50],因此預防寄生蟲的感染尤為重要。目前寄生蟲的常規控制措施包括消滅傳染源、切斷傳播途徑以及保護易感宿主[51]。利用scRNA-seq技術能夠從細胞層面表征寄生蟲的發育繁殖方式,從而開發寄生蟲相關疫苗。
錐蟲屬(Trypanosoma)隸屬于原生動物門肉鞭動物亞門動鞭毛綱動體目錐蟲科,本屬動物統稱錐蟲[52]。研究顯示,布氏錐蟲(Trypanosoma brucei)必須在采采蠅的唾液腺中發育成感染哺乳動物的后循環型錐鞭毛體,才能傳播到新的宿主中[53]。Hutchinson等[54]通過inDrop技術表征了布氏錐蟲在唾液腺中的細胞異質性。Vigneron等[55]利用scRNA-seq技術從受布氏錐蟲感染的唾液腺中分離出2 045個不同布氏錐蟲個體的寄生蟲轉錄組,根據轉錄活性和特征以及表面被膜蛋白的表達,將轉錄組聚類為布氏錐蟲不同生長發育階段的3個細胞類群,通過在不同的細胞類群中尋找編碼表面相關蛋白的高豐度差異表達基因,發現了存在于后循環期蟲體細胞表面的一種顯著的非變異外殼蛋白(SGM1.7),并且抗SGM1.7抗體的研究結果顯示,該蛋白涉及B細胞抗體介導的免疫反應,該反應可以在哺乳動物感染的初始階段干擾寄生蟲的存活或分化,提示該蛋白具有作為亞單位疫苗抗原的潛力。
此外,血吸蟲在入侵宿主過程中,也會經歷從自由生活、高度活動的尾蚴到成蟲寄生形式的主要生理和形態轉變[56]。Wang等[57] 利用scRNA-seq技術分析了從曼氏血吸蟲尾蚴和童蟲階段分離的細胞,共鑒定了四種干細胞類型,包括κ細胞、δ細胞、φ細胞和ε細胞,其中ε細胞特異性表達的基因eledh在尾蚴中檢測不到,但在幼蟲干細胞中是最豐富的轉錄本之一。研究表明,eledh是血吸蟲中最早發現的種系標志物,并且生殖細胞可能來源于發育早期的ε細胞,這種早期增殖的細胞可能有助于寄生蟲生長和成熟所需的組織重塑。這項研究為理解驅動血吸蟲繁殖和長期存活的基本機制邁出了重要一步,通過表征此處定義的干細胞群體在血吸蟲傳播、生殖發育和存活中的作用,有望研發減少寄生蟲荷蟲量,抑制寄生蟲在宿主體內發育的新型疫苗,從而有效控制血吸蟲的廣泛傳播。
3.3 在寄生蟲病藥物研究中的應用
scRNA-seq技術在藥物研究中的應用包括藥物靶點的發現、高通量藥物的篩選、藥物不良反應機制和耐藥機制的揭示等[58]。瘧疾是由瘧原蟲引起的一種寄生蟲病,宿主經按蚊叮咬或輸入帶瘧原蟲者的血液而感染該病,其中惡性瘧原蟲感染容易引發膿毒癥[59]。Rawat等[60]對惡性瘧原蟲在高溫(相當于發燒)的生理應激條件和對照條件下進行了scRNA-seq分析,發現在不同條件下,雖然寄生蟲在形態上是同步的,但它們表現出轉錄組上的異質性,其中3組細胞類群在應激條件的刺激下上調了許多配子體標記和調節因子的表達,它們可能與惡劣條件下瘧原蟲的脅迫適應相關。由于應激反應是惡性瘧原蟲產生耐藥性的重要決定因素[61],因此這項研究對了解瘧原蟲耐藥性的產生具有重要意義。青蒿琥酯(artesunate, ART)是一種抗瘧疾藥物,He等[62]利用scRNA-seq技術,全面描述了小鼠膿毒癥模型中肝臟的生物學特性,并通過對scRNA-seq數據的分析,驗證了ART治療膿毒癥后對肝功能恢復的影響,發現ART治療后可以顯著降低小鼠血清中內毒素的含量、改善肝內皮細胞的損傷、有效抑制巨噬細胞的募集以及減輕淋巴細胞免疫功能障礙,提高了96 h內小鼠的存活率,從而揭示了ART有效治療膿毒癥小鼠的部分作用機制。Zheng等[63]通過scRNA-seq技術發現,將ART與化療或免疫治療藥物聯用,能夠干預乳腺癌中腫瘤相關成纖維細胞和癌細胞,獲得更好的治療效果,從而發現該藥物的不同適應癥。
scRNA-seq技術在藥物篩選方面也具有一定優勢,2018年,Ye等[64]結合scRNA-seq技術,開發了數字核糖核酸與基因突變測序平臺(DRUG-seq),該平臺能夠高通量的篩選藥物,同時還可以用于分析基因敲除細胞,以揭示基因和化合物的功能。最近,Xie等[65]基于scRNA-seq技術報道了組合擾動測序技術(CP-seq),它可以同時以高度多重的方式干擾藥物組合的細胞,并執行單細胞轉錄組分析,以此來表征組合藥物對單細胞的基因擾動情況,其在尋找新的有效藥物組合方面具有巨大潛力。總之,利用scRNA-seq技術能夠從細胞層面進一步揭示寄生蟲的耐藥機制以及抗寄生蟲藥物在治療過程中的作用機制,用于輔助新型化合物機制研究的同時,豐富藥物的臨床適應癥范圍。另外,scRNA-seq技術大規模、自動化、高通量、低成本的特點也為抗寄生蟲藥物篩選提供新的技術路線,有助于準確、高效和特異地篩選藥物,加速藥物研發進程,為治療寄生蟲病提供有效的技術支撐。
3.4 在寄生蟲病治療靶點鑒定中的應用
對于具有移行功能或致病性較強的寄生蟲,大部分患者目前無法得到根治,因此開發新型治療手段具有重要意義。棘球蚴病主要包括囊型棘球蚴病(cystic echinococcosis, CE)和泡型棘球蚴病(alveolar echinococcosis, AE),其中AE是由多房棘球絳蟲引起的一種廣泛傳播的人畜共患病,寄生蟲通過消化道后,主要寄生在肝臟,在易感宿主中,中絳期的幼蟲可在患者體內像腫瘤樣生長發育和浸潤性增殖,因此該病又稱為“蟲癌”[66]。Yarahmadov等[67]利用scRNA-seq技術評估了多房棘球絳蟲蟲卵經口感染小鼠肝臟中免疫細胞的異質性,從感染后3、10、21 d以及對照小鼠肝臟中提取CD3+ T細胞進行scRNA-seq,最終獲得了5種不同類型T細胞。并發現自然殺傷T細胞對多房棘球絳蟲感染反應的效應機制主要與IFN-γ、TNF、IL-7和IL-18相關的信號傳導、細胞毒性途徑以及表達顆粒酶b和殺傷細胞凝集素的受體有關。而免疫抑制相關的基因包括免疫檢查點(PD-1/PD-L1)和嘌呤能信號(ENTPD1/ADORA2A),它們在感染過程中也極有可能通過調節自然殺傷T細胞的功能從而將慢性疾病期間的免疫抑制轉化為促炎反應,抑制寄生蟲生長,這些結果為AE患者免疫調節治療靶標的選擇提供了依據。
由細粒棘球絳蟲引起的CE呈全球性分布,其在中國西北部、中東、北非和南美洲的牧區具有高感染率[68]。但大部分CE患者會錯過最佳手術時期,對于晚期病例,姑息性手術和抗感染治療均不理想[69],因此亟待開發新型治療手段。Yasen等[70]對四名CE患者的外周血、病灶周圍肝組織和鄰近正常肝組織的免疫細胞進行scRNA-seq,共鑒定了23種細胞類群,發現固有淋巴細胞的基因表達存在顯著差異,其在病灶周圍肝組織中差異調控的基因主要富集于白細胞活化、toll樣受體信號通路、NF-κB信號通路和免疫系統過程負調控方面。此外,潛在的抑制性免疫檢查點基因NKG2A及其配體HLA-E在病灶中的表達顯著高于相應的鄰近正常肝組織,證明了該免疫檢查點在CE病變微環境中的重要抑制作用。隨后,Jiang等[71]也利用scRNA-Seq揭示了人體CE病變微環境中浸潤免疫細胞的轉錄異質性,首次發現CE患者后期特異性出現的巨噬細胞群,并通過高表達基因將其鑒定為SPP1+巨噬細胞。該細胞類型高表達許多免疫抑制和促血管生成基因,與免疫微環境中的腫瘤相關巨噬細胞群體相似,有助于腫瘤細胞免疫逃逸并促進腫瘤血管生成。這些在CE發展過程中對免疫調節反應具有重要功能的分子,均有望成為CE的潛在治療靶點。
3.5 對寄生蟲病診斷技術的推進
寄生蟲病的診斷需要高度敏感和特異性的檢查方法,其中最可靠的方法是檢測和鑒定到感染的病原體,而寄生蟲的鑒定通常涉及其流行病學,以及對蟲種和亞種的區分[72]。臨床醫學上,僅僅根據流行病學和臨床癥狀很難確診某種寄生蟲病,需要借助一些實驗室診斷技術才能準確判斷病原,目前臨床上常用的實驗室診斷方法有常規形態學診斷、血清學診斷和分子生物學診斷。利用scRNA-seq技術也能在診斷寄生蟲病方面發揮作用,一方面,scRNA-seq技術能夠發現寄生蟲中特異性表達的基因以及具有異質性的細胞類型,例如Reid等[73]利用scRNA-seq技術揭示了瘧原蟲中隱藏的轉錄變異,發現一些細胞似乎具有在無性細胞和配子細胞之間的中間轉錄組。Louradour等[74]通過scRNA-seq技術鑒定了利什曼原蟲在基因轉錄上具有獨特性的前鞭毛體群體,這些群體的數量在γ輻射后的每個親本系中顯著增加,并且其特定的減數分裂基因同源基因被發現上調,其中包括祖先配子HAP2。這些研究為寄生蟲免疫學診斷和核酸診斷提供了潛在靶點。另一方面,Xue等[75]利用scRNA-seq技術對5 400多個弓形蟲的速殖子和緩殖子兩個階段進行了研究,構建了第一個全面的弓形蟲無性發育階段的單細胞圖譜,揭示了AP2IX-1是一種新的轉錄因子,結合瞬時表達試驗,發現AP2IX-1在分化過程中參與了表面抗原譜系的重塑,并且可能在控制弓形蟲性別分化中起關鍵作用。同時發現每種弓形蟲的基因組中都存在120多個與SAG1相關的基因,它們產生的一整套蛋白質可能會“隱藏”這些寄生蟲,使其不被免疫系統發現。這些結果表明寄生蟲在感染宿主期間具有免疫逃避機制,在大規模測序中,這些免疫逃避和其他功能相關分子無法全部檢測和鑒定,但scRNA-seq技術具有更高的敏感性,能夠捕捉每個細胞中的轉錄組信息,從而特異的提供寄生蟲細胞中基因表達的詳細信息,預計未來宿主細胞和寄生蟲的單細胞共轉錄測序將成為進一步了解并診斷或鑒別寄生蟲亞種的潛在有效方法。
3.6 在寄生蟲親緣關系分析和進化研究中的應用
理解寄生蟲親緣關系對于生物學、醫學和生態學等領域都具有重要的意義,有助于認識不同寄生蟲之間的演化關系,從而更好地進行分類和建立系統發育樹,進一步揭示物種之間的演化歷史和彼此之間的親緣關系。PlanMine提供了當前扁形動物門可利用的序列庫,Rozanski等[76]為其添加了來自各種其他扁蟲的單細胞轉錄組數據,并提供了一個系統發育的交互網絡,為扁蟲生物學的比較分析提供資源,以期實現有意義的物種間比較。
頂復門(Apicomplexa)是原生動物門中的一門,包括形態和生態上不同的原生動物,例如隱孢子蟲、球蟲和血孢子蟲等[77]。Janouskovec等[78]利用scRNA-seq研究了十種不同的頂復門寄生蟲,分析發現頂復門寄生蟲沒有近似的共同祖先,因此從進化的角度來看,它們不是一個自然的群體,相反,它們相似的外觀和生活方式至少在三個獨立的時期內進化。并且在后續的研究發現,至少有三個頂復門譜系喪失了它們的隱蔽葉綠體,該研究結果為葉綠體在生命演化中的重要性提供了新的觀點。Mathur等[79]使用單細胞測序表征了部分研究很少的頂復門寄生蟲的基因組和轉錄組,發現先前在頂復門中分類的兩個屬(Piridium和Platyproteum),能夠在系統發育分析中形成單獨的分支譜系,兩者都保留了具有與頂復門生物相似的基因組和代謝特征的隱性質體,該譜系對光合作用的趨同性喪失和向寄生轉變,導致多個譜系中出現了表面上相似的動物寄生蟲。
隨后,Mathur等[80]對未被充分研究的gregarine譜系進行了scRNA-seq,構建了一個包含Apicomplexa、Chrompodellids和Squirmida(ACS分支)可用測序數據集的系統發育樹,利用這些數據集,Mathur團隊詳細檢查了弓形蟲空間蛋白質組學顯示在頂端體中的162種蛋白質的進化分布,證實了在光合作用喪失后保留的整體代謝途徑具有趨同趨勢,揭示了特定譜系中質體減少程度的差異,同時發現質體基因組的丟失很常見,并且意外地發現了許多譜系和物種特異性的質體蛋白,這表明進化創新和新功能的存在可能賦予這些神秘的細胞器尚未發現的新功能和代謝能力。
此外,Lahr等[81]提出了表殼目(Arcellinida)的全面系統發育重建,通過scRNA-seq從19個具有代表性的有殼變形蟲分類群中獲得轉錄組數據,由此生成的進化樹得到強有力的支持,首次揭示了該群體最深的譜系。利用數據集,Lahr團隊進行了祖先形態的重建,得到了與現存新元古代瓶形微體化石非常相似的假設祖先。這些結果為表殼目創造了一個堅實的系統發育骨干,并刺激了對該群體相關化石記錄的重新解釋,可能闡明真核生物早期進化中的關鍵事件。Kang等[82]利用scRNA-seq技術對61個變形蟲類群進行采樣,分析顯示出變形蟲存在兩個主要分支(Discosea和Tevosa),認為變形蟲的主要宏觀進化模式是由多階段生命周期(包括鞭毛、有性和子實體)的同源性特征的平行喪失所導致,而不是獨立獲得的相似特征。總之,寄生蟲親緣關系的研究為人們提供了深入了解生命演化、生物進化以及生態系統結構功能的機會,為相關領域的科研和實踐提供了基礎。
4 小結與展望
scRNA-seq方法自2009年首次引入以來,為揭示組織細胞組成、細胞異質性、轉錄組動力學和基因之間的調控關系等開辟了一條新的途徑,隨著高效和低成本技術的出現,scRNA-seq技術的商業化平臺已經足以建立數千個細胞的測序文庫,單細胞技術至此已經作為標準程序使用,廣泛應用于生物學、醫學、寄生蟲學和臨床等研究領域。然而,目前單細胞轉錄組技術仍然面臨若干重大挑戰:首先,大部分測序技術通過poly(A)尾構建文庫,其主要檢測局限于mRNA和部分帶ploy(A)尾的長鏈非編碼RNA,對于大量存在的非ploy(A)尾長鏈非編碼RNA的檢測還存在一定困難;第二,scRNA-seq技術基于組織解離進行,捕獲單個細胞轉錄組的同時會丟失其空間位置信息,而揭示組織細胞的空間位置分布對于疾病理解具有重要意義,因此亟需發展單細胞與空間多組學聯合研究方法;第三,單細胞測序數據通常來自于多次試驗,由于樣本、實驗操作和技術平臺等因素的不同,測序數據總是存在差異性。因此在進行數據分析時,去批次效應具有重要意義,但當前常用的評測批次效應的定性或者定量算法工具均存在一定局限性。
盡管如此,單細胞轉錄組學技術仍是發現新的診斷或治療靶點強有力的工具,其在寄生蟲研究領域中揭示了部分寄生蟲的生長發育圖譜、探析了同屬寄生蟲不同的種屬基因組、表征了宿主和寄生蟲如何相互適應的基因表達譜、信號傳導通路和代謝途徑。總而言之,隨著科技及各種單細胞組學工具的發展,scRNA-seq技術能夠更好地應用于寄生蟲學研究,為理解寄生蟲發育與致病機制,防治寄生蟲病提供更好的技術支撐。
參考文獻(References):
[1] CHAMBERS D C, CAREW A M, LUKOWSKI S W, et al. Transcriptomics and single-cell RNA-sequencing[J]. Respirology, 2019, 24(1):29-36.
[2] MANZONI C, KIA D A, VANDROVCOVA J, et al. Genome, transcriptome and proteome:the rise of omics data and their integration in biomedical sciences[J]. Brief Bioinform, 2018, 19(2):286-302.
[3] EBERWINE J, YEH H, MIYASHIRO K, et al. Analysis of gene expression in single live neurons[J]. Proc Natl Acad Sci U S A, 1992, 89(7):3010-3014.
[4] TANG F C, BARBACIORU C, WANG Y Z, et al. mRNA-Seq whole-transcriptome analysis of a single cell[J]. Nat Methods, 2009, 6(5):377-382.
[5] JOVIC D, LIANG X, ZENG H, et al. Single-cell RNA sequencing technologies and applications:A brief overview[J]. Clin Transl Med, 2022, 12(3):e694.
[6] 楊富升, 古小彬. 近十年PCR技術在寄生蟲病診斷中的應用[J]. 畜牧獸醫學報, 2023, 54(8):3183-3194.
YANG F S, GU X B. A review on applications of PCR technology in the diagnosis of parasitic diseases in the past 10 years[J]. Acta Veterinaria et Zootechnica Sinica, 2023, 54(8):3183-3194. (in Chinese)
[7] LI P Y, SARFATI D N, XUE Y, et al. Single-cell analysis of Schistosoma mansoni identifies a conserved genetic program controlling germline stem cell fate[J]. Nat Commun, 2021, 12(1):485.
[8] FENG M, ZHANG Y H, ZHOU H, et al. Single-cell RNA sequencing reveals that the switching of the transcriptional profiles of cysteine-related genes alters the virulence of Entamoeba histolytica[J]. mSystems, 2020, 5(6):e01095-20.
[9] RUBERTO A A, BOURKE C, MERIENNE N, et al. Single-cell RNA sequencing reveals developmental heterogeneity among Plasmodium berghei sporozoites[J]. Sci Rep, 2021, 11(1):4127.
[10] OLSEN T K, BARYAWNO N. Introduction to Single-cell RNA sequencing[J]. Curr Protoc Mol Biol, 2018, 122(1):e57.
[11] PAIK D T, CHO S, TIAN L, et al. Single-cell RNA sequencing in cardiovascular development, disease and medicine[J]. Nat Rev Cardiol, 2020, 17(8):457-473.
[12] ZIEGENHAIN C, VIETH B, PAREKH S, et al. Comparative analysis of Single-cell RNA sequencing methods[J]. Mol Cell, 2017, 65(4):631-643. e4.
[13] HASHIMSHONY T, WAGNER F, SHER N, et al. CEL-Seq:single-cell RNA-Seq by multiplexed linear amplification[J]. Cell Rep, 2012, 2(3):666-673.
[14] HASHIMSHONY T, SENDEROVICH N, AVITAL G, et al. CEL-Seq2:sensitive highly-multiplexed single-cell RNA-Seq[J]. Genome Biol, 2016, 17:77.
[15] JAITIN D A, KENIGSBERG E, KEREN-SHAUL H, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types[J]. Science, 2014, 343(6172):776-779.
[16] KEREN-SHAUL H, KENIGSBERG E, JAITIN D A, et al. MARS-seq2. 0:an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing[J]. Nat Protoc, 2019, 14(6):1841-1862.
[17] RAMSK?LD D, LUO S J, WANG Y C, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells[J]. Nat Biotechnol, 2012, 30(8):777-782.
[18] PICELLI S, BJ?RKLUND K, FARIDANI O R, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells[J]. Nat Methods, 2013, 10(11):1096-1098.
[19] HAGEMANN-JENSEN M, ZIEGENHAIN C, CHEN P, et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3[J]. Nat Biotechnol, 2020, 38(6):708-714.
[20] HAGEMANN-JENSEN M, ZIEGENHAIN C, SANDBERG R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress[J]. Nat Biotechnol, 2022, 40(10):1452-1457.
[21] SOUMILLON M, CACCHIARELLI D, SEMRAU S, et al. Characterization of directed differentiation by high-throughput single-cell RNA-Seq[J]. bioRxiv, 2014:003236.
[22] MACOSKO E Z, BASU A, SATIJA R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets[J]. Cell, 2015, 161(5):1202-1214.
[23] KLEIN A M, MAZUTIS L, AKARTUNA I, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells[J]. Cell, 2015, 161(5):1187-1201.
[24] ZHENG G X Y, TERRY J M, BELGRADER P, et al. Massively parallel digital transcriptional profiling of single cells[J]. Nat Commun, 2017, 8:14049.
[25] CHEN W Z, GUILLAUME-GENTIL O, RAINER P Y, et al. Live-seq enables temporal transcriptomic recording of single cells[J]. Nature, 2022, 608(7924):733-740.
[26] HAN X P, WANG R Y, ZHOU Y C, et al. Mapping the mouse cell atlas by microwell-seq[J]. Cell, 2018, 172(5):1091-1107. e17.
[27] GIERAHN T M, WADSWORTH II M H, HUGHES T K, et al. Seq-Well:portable, low-cost RNA sequencing of single cells at high throughput[J]. Nat Methods, 2017, 14(4):395-398.
[28] 熊和麗, 沙 茜, 劉韶娜, 等. 單細胞轉錄組測序技術在動物上的應用研究[J]. 生物技術通報, 2022, 38(3):226-233.
XIONG H L, SHA Q, LIU S N, et al. Application of single-cell transcriptome sequencing in animals. [J]. Biotechnology Bulletin, 2022, 38(3):226-233. (in Chinese)
[29] ZHANG X N, LI T Q, LIU F, et al. Comparative analysis of droplet-based ultra-high-throughput single-cell rna-seq systems[J]. Mol Cell, 2019, 73(1):130-142. e5.
[30] MEISTER A, GABI M, BEHR P, et al. FluidFM:combining atomic force microscopy and nanofluidics in a universal liquid delivery system for single cell applications and beyond[J]. Nano Lett, 2009, 9(6):2501-2507.
[31] PICELLI S, FARIDANI O R, BJ?RKLUND K, et al. Full-length RNA-seq from single cells using Smart-seq2[J]. Nat Protoc, 2014, 9(1):171-181.
[32] HORVATH R. Single-cell temporal transcriptomics from tiny cytoplasmic biopsies[J]. Cell Rep Methods, 2022, 2(10):100319.
[33] TANG Q K, LI W J, HUANG J, et al. Single-cell sequencing analysis of peripheral blood in patients with moyamoya disease[J]. Orphanet J Rare Dis, 2023, 18(1):174.
[34] DOBIN A, DAVIS C A, SCHLESINGER F, et al. STAR:ultrafast universal RNA-seq aligner[J]. Bioinformatics, 2013, 29(1):15-21.
[35] YOU Y, TIAN L Y, SU S A, et al. Benchmarking UMI-based single-cell RNA-seq preprocessing workflows[J]. Genome Biol, 2021, 22(1):339.
[36] ENGSTR?M P G, STEIJGER T, SIPOS B, et al. Systematic evaluation of spliced alignment programs for RNA-seq data[J]. Nat Methods, 2013, 10(12):1185-1191.
[37] CHEN G, NING B T, SHI T L. Single-cell RNA-seq technologies and related computational data analysis[J]. Front Genet, 2019, 10:317.
[38] SLOVIN S, CARISSIMO A, PANARIELLO F, et al. Single-cell RNA sequencing analysis:a step-by-step overview[J]. Methods Mol Biol, 2021, 2284:343-365.
[39] BUTLER A, HOFFMAN P, SMIBERT P, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species[J]. Nat Biotechnol, 2018, 36(5):411-420.
[40] CAO J Y, SPIELMANN M, QIU X J, et al. The single-cell transcriptional landscape of mammalian organogenesis[J]. Nature, 2019, 566(7745):496-502.
[41] CHEN Q X, YIN Q J, SONG J X, et al. Identification of monocyte-associated genes as predictive biomarkers of heart failure after acute myocardial infarction[J]. BMC Med Genomics, 2021, 14(1):44.
[42] 趙琴平, 董惠芬, 蔣明森. 關于寄生蟲病防治研究的幾點思考[J]. 中國血吸蟲病防治雜志, 2013, 25(6):564-569, 589.
ZHAO Q P, DONG H F, JIANG M S. Views for research development of control of parasitic diseases[J]. Chinese Journal of Schistosomiasis Control, 2013, 25(6):564-569, 589. (in Chinese)
[43] TINTORI S C, NISHIMURA E O, GOLDEN P, et al. A transcriptional lineage of the early C. elegans embryo[J]. Dev Cell, 2016, 38(4):430-444.
[44] LORENZO R, ONIZUKA M, DEFRANCE M, et al. Combining single-cell RNA-sequencing with a molecular atlas unveils new markers for Caenorhabditis elegans neuron classes[J]. Nucleic Acids Res, 2020, 48(13):7119-7134.
[45] TAYLOR S R, SANTPERE G, WEINREB A, et al. Molecular topography of an entire nervous system[J]. Cell, 2021, 184(16):4329-4347. e23.
[46] FINCHER C T, WURTZEL O, DE HOOG T, et al. Cell type transcriptome atlas for the planarian Schmidtea mediterranea[J]. Science, 2018, 360(6391):eaaq1736.
[47] ZENG A, LI H, GUO L H, et al. Prospectively isolated tetraspanin+ neoblasts are adult pluripotent stem cells underlying planaria regeneration[J]. Cell, 2018, 173(7):1593-1608. e20.
[48] WURTZEL O, COTE L E, POIRIER A, et al. A generic and cell-type-specific wound response precedes regeneration in planarians[J]. Dev Cell, 2015, 35(5):632-645.
[49] SONG L G, ZENG X D, LI Y X, et al. Imported parasitic diseases in mainland China:current status and perspectives for better control and prevention[J]. Infect Dis Poverty, 2018, 7(1):78.
[50] BAHK Y Y, SHIN E H, CHO S H, et al. Prevention and control strategies for parasitic infections in the Korea centers for disease control and prevention[J]. Korean J Parasitol, 2018, 56(5):401-408.
[51] 葉美瓊. 寄生蟲病的危害及防治對策[J]. 今日畜牧獸醫, 2021, 37(5):88-89.
YE M Q. The harm and prevention strategies of parasitic diseases[J]. Today Animal Husbandry and Veterinary Medicine, 2021, 37(5):88-89.
[52] LSCHER A, DE KONING H P, M?SER P. Chemotherapeutic strategies against Trypanosoma brucei:drug targets vs." drug targeting[J]. Curr Pharm Des, 2007, 13(6):555-567.
[53] MATETOVICI I, CALJON G, VAN DEN ABBEELE J. Tsetse fly tolerance to T. brucei infection:transcriptome analysis of trypanosome-associated changes in the tsetse fly salivary gland[J]. BMC Genomics, 2016, 17(1):971.
[54] HUTCHINSON S, FOULON S, CROUZOLS A, et al. The establishment of variant surface glycoprotein monoallelic expression revealed by single-cell RNA-seq of Trypanosoma brucei in the tsetse fly salivary glands[J]. PLoS Pathog, 2021, 17(9):e1009904.
[55] VIGNERON A, O’NEILL M B, WEISS B L, et al. Single-cell RNA sequencing of Trypanosoma brucei from tsetse salivary glands unveils metacyclogenesis and identifies potential transmission blocking antigens[J]. Proc Natl Acad Sci U S A, 2020, 117(5):2613-2621.
[56] SORIA C L D, LEE J, CHONG T, et al. Single-cell atlas of the first intra-mammalian developmental stage of the human parasite Schistosoma mansoni[J]. Nat Commun, 2020, 11(1):6411.
[57] WANG B, LEE J, LI P Y, et al. Stem cell heterogeneity drives the parasitic life cycle of Schistosoma mansoni[J]. eLife, 2018, 7:e35449.
[58] 汪茂林, 楊洪軍. 單細胞轉錄組測序技術在藥物研究中的應用[J]. 藥學學報, 2023, 58(9):2551-2559.
WANG M L, YANG H J. Single cell RNA sequencing technology applicated for drug discovery[J]. Acta Pharmaceutica Sinica, 2023, 58(9):2551-2559. (in Chinese)
[59] WHITE N J. Erratum to:Malaria parasite clearance[J]. Malar J, 2017, 16(1):194.
[60] RAWAT M, SRIVASTAVA A, JOHRI S, et al. Single-cell RNA sequencing reveals cellular heterogeneity and stage transition under temperature stress in synchronized Plasmodium falciparum cells[J]. Microbiol Spectr, 2021, 9(1):e0000821.
[61] ROCAMORA F, ZHU L, LIONG K Y, et al. Oxidative stress and protein damage responses mediate artemisinin resistance in malaria parasites[J]. PLoS Pathog, 2018, 14(3):e1006930.
[62] HE X L, CHEN J Y, FENG Y L, et al. Single-cell RNA sequencing deciphers the mechanism of sepsis-induced liver injury and the therapeutic effects of artesunate[J]. Acta Pharmacol Sin, 2023, 44(9):1801-1814.
[63] ZHENG D D, ZHOU J, QIAN L, et al. Biomimetic nanoparticles drive the mechanism understanding of shear-wave elasticity stiffness in triple negative breast cancers to predict clinical treatment[J]. Bioact Mater, 2023, 22:567-587.
[64] YE C Y, HO D J, NERI M, et al. DRUG-seq for miniaturized high-throughput transcriptome profiling in drug discovery[J]. Nat Commun, 2018, 9(1):4307.
[65] XIE R, LIU Y, WANG S Y, et al. Combinatorial perturbation sequencing on single cells using microwell-based droplet random pairing[J]. Biosens Bioelectron, 2023, 220:114913.
[66] AUTIER B, MANUEL C, LUNDSTROEM-STADELMANN B, et al. Endogenous IL-33 accelerates metacestode growth during late-stage alveolar echinococcosis[J]. Microbiol Spectr, 2023, 11(2):e04239-22.
[67] YARAHMADOV T, WANG J H, SANCHEZ-TALTAVULL D, et al. Primary infection by E. multilocularis induces distinct patterns of cross talk between hepatic natural killer T Cells and regulatory T cells in mice[J]. Infect Immun, 2022, 90(8):e00174-22.
[68] WEN H, VUITTON L, TUXUN T, et al. Echinococcosis:advances in the 21st century[J]. Clin Microbiol Rev, 2019, 32(2):e00075-18.
[69] JUNGHANSS T, DA SILVA A M, HORTON J, et al. Clinical management of cystic echinococcosis:state of the art, problems, and perspectives[J]. Am J Trop Med Hyg, 2008, 79(3):301-311.
[70] YASEN A, SUN W, AINI A, et al. Single-cell RNA sequencing reveals the heterogeneity of infiltrating immune cell profiles in the hepatic cystic echinococcosis microenvironment[J]. Infect Immun, 2021, 89(12):e0029721.
[71] JIANG X F, ZHANG X F, JIANG N, et al. The single-cell landscape of cystic echinococcosis in different stages provided insights into endothelial and immune cell heterogeneity[J]. Front Immunol, 2022, 13:1067338.
[72] AMBROSIO R E, DE WAAL D T. Diagnosis of parasitic disease[J]. Rev Sci Tech, 1990, 9(3):759-778.
[73] REID A J, TALMAN A M, BENNETT H M, et al. Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites[J]. eLife, 2018, 7:e33105.
[74] LOURADOUR I, FERREIRA T R, DUGE E, et al. Stress conditions promote Leishmania hybridization in vitro marked by expression of the ancestral gamete fusogen HAP2 as revealed by single-cell RNA-seq[J]. eLife, 2022, 11:e73488.
[75] XUE Y, THEISEN T C, RASTOGI S, et al. A single-parasite transcriptional atlas of Toxoplasma Gondii reveals novel control of antigen expression[J]. eLife, 2020, 9:e54129.
[76] ROZANSKI A, MOON H, BRANDL H, et al. PlanMine 3. 0-improvements to a mineable resource of flatworm biology and biodiversity[J]. Nucleic Acids Res, 2019, 47(D1):D812-D820.
[77] VOTYPKA J, MODRY D, OBORNíK M, et al. Apicomplexa[M]∥ARCHIBALD J M, SIMPSON A G B, SLAMOVITS C H. Handbook of the Protists. 2nd ed. New York:Springer, 2017:567-624.
[78] JANOU?KOVEC J, PASKEROVA G G, MIROLIUBOVA T S, et al. Apicomplexan-like parasites are polyphyletic and widely but selectively dependent on cryptic plastid organelles[J]. eLife, 2019, 8:e49662.
[79] MATHUR V, KOLíSKO M, HEHENBERGER E, et al. Multiple independent origins of Apicomplexan-like parasites[J]. Curr Biol, 2019, 29(17):2936-2941. e5.
[80] MATHUR V, SALOMAKI E D, WAKEMAN K C, et al. Reconstruction of plastid proteomes of apicomplexans and close relatives reveals the major evolutionary outcomes of cryptic plastids[J]. Mol Biol Evol, 2023, 40(1):msad002.
[81] LAHR D J G, KOSAKYAN A, LARA E, et al. Phylogenomics and morphological reconstruction of arcellinida testate amoebae highlight diversity of microbial eukaryotes in the neoproterozoic[J]. Curr Biol, 2019, 29(6):991-1001. e3.
[82] KANG S, TICE A K, SPIEGEL F W, et al. Between a pod and a hard test:the deep evolution of amoebae[J]. Mol Biol Evol, 2017, 34(9):2258-2270.
(編輯 白永平)
猜你喜歡
職工法律天地·下半月(2016年10期)2016-11-30 11:52:57
商情(2016年40期)2016-11-28 11:28:07
商(2016年32期)2016-11-24 17:39:41
科技資訊(2016年18期)2016-11-15 18:05:53
考試周刊(2016年84期)2016-11-11 23:57:34
科技視界(2016年18期)2016-11-03 22:51:40
體育時空(2016年8期)2016-10-25 18:02:39
現代經濟信息(2016年19期)2016-10-20 17:46:29
中國科技博覽(2016年18期)2016-10-19 10:30:11
中國市場(2016年36期)2016-10-19 04:31:23
