杜仲片質量標準研究
2018-03-05 05:45:48林向前
中國藥業 2018年4期
林向前
(福建省漳州市中醫院,福建 漳州 363000)
杜仲片是我院骨傷科經典傳統中藥制劑,可補腎強腰、活血化瘀,適用于急慢性腰部損傷、關節酸痛等腎虛血瘀證,50多年的臨床應用證明,該藥療效確切且未見不良反應[1]。為控制其質量,筆者參考文獻[2],建立了杜仲片的質量標準。現報道如下。
1 儀器與試藥
儀器:Agilent 1260型高效液相色譜儀,包括Agilent-G1311C型四元泵,Agilent G1329B型真空脫氣器,Agilent-G1315D型二極管陣列檢測器,Chemstation B.04.02型化學工作站[3](美國安捷倫公司);KQ-250B 型超聲清洗機(昆山超聲儀器有限公司);Mettler Toledo EL2401型電子天平(瑞士梅特勒-托利多公司)。
試藥:芍藥苷對照品(批號為 110736-201136,含量以96.0%計,使用前無需處理),當歸對照藥材(批號為 927-200110),丹皮酚對照品(批號為 0708-9704),延胡索乙素對照品(批號為0726-200208),均購自中國食品藥品檢定研究院;杜仲片(批號分別為20150701,20150901,20151101),杜仲片缺味陰性對照品,均由本院制劑中心提供;乙腈為色譜純,水為重蒸餾水,其他試劑均為分析純。
2 方法與結果
2.1 一般質量控制
性狀:本品為褐黑色片;氣微辛,味微苦。
檢查:按2015年版《中國藥典(四部)》通則0101片劑項下有關規定,對3批樣品重量差異、崩解時限、微生物限度進行檢查,結果均符合規定[4]。
2.2 定性鑒別
當歸:取杜仲片20片,搗碎成細粉,加乙醚40 mL,超聲處理5 min,靜置2 h,濾過,濾液蒸發干燥,干燥物加入乙酸乙酯1 mL,充分攪拌使其溶解,作為供試品溶液。另取當歸對照藥材1 g,按上述方法制備對照藥材溶液[5]。同法制備缺當歸的陰性對照品溶液。按2015年版《中國藥典(四部)》通則0502薄層色譜法[4]試驗,吸取上述3種溶液各6 μL,分別點于同一硅膠G薄層板上,以石油醚(60~90℃)-乙酸乙酯(50∶1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視[6]。供試品溶液色譜中,在與對照藥材溶液色譜相應位置上顯相同顏色的熒光斑點,陰性對照無干擾(圖1 A)。
牡丹皮:取當歸鑒別項下的供試品溶液,作為供試品溶液。另取丹皮酚對照品,加乙酸乙酯制成每1 mL含1 mg的溶液,作為對照品溶液。同法制備缺牡丹皮飲片的陰性對照品溶液[5]。照2015年版《中國藥典(四部)》通則0502薄層色譜法[4]試驗,吸取上述3種溶液各8 μL,分別點于同一硅膠G薄層板上,以環己烷-乙酸乙酯-冰醋酸(10 ∶1 ∶0.1)為展開劑,展開,取出,晾干,置紫外光燈(254 nm)下檢視[6]。供試品溶液色譜中,在與對照品溶液色譜相應位置上顯相同顏色的熒光斑點,陰性對照無干擾(圖1 B)。
延胡索:取杜仲片30片,搗碎成細粉,加甲醇50 mL,超聲處理20 min,濾過,濾液蒸發干燥,干燥物加水15 mL攪拌使其溶解,用氨水調pH至9~10,用乙醚提取2次,每次15 mL,合并乙醚液,揮干,殘渣加乙酸乙酯1 mL充分攪拌使溶解,作為供試品溶液[5]。另取延胡索乙素對照品,加甲醇制成每1 mL含1 mg的溶液,作為對照品溶液。同法制備缺醋延胡索飲片的陰性對照品溶液。照2015年版《中國藥典(四部)》通則0502薄層色譜法[4]試驗,吸取上述3種溶液各8 μL,分別點于同一硅膠 G 薄層板上,以正己烷 - 氯仿 - 甲醇(7.5∶4∶1)為展開劑,展開,取出,晾干,置碘蒸氣熏至斑點顯色清晰,在空氣中揮盡板上吸附的碘后,置紫外光燈(365 nm)下檢視[6]。供試品溶液色譜中,在與對照品溶液色譜相應位置上顯相同顏色的熒光斑點,陰性對照無干擾(圖1 C)。

圖1 杜仲片薄層色譜圖
2.3 芍藥苷含量測定
2.3.1 色譜條件及系統適用性試驗
色譜柱:Lichrospher-C18柱(250 mm ×4.6 mm,5 μm);流動相:乙腈 - 0.1% 磷酸溶液(13 ∶87);流速:1.0 mL /min;檢測波長:230 nm;柱溫:30 ℃ ;進樣量:10 μL。在此色譜條件下,芍藥苷達到基線分離(芍藥苷色譜峰與相鄰色譜峰之間的分離度大于1.50)[7],理論板數按芍藥苷峰計應不低于2 000,詳見圖2。
2.3.2 溶液制備
取杜仲片10片,搗碎成細粉,稱取約0.5 g,精密稱定,放入帶有瓶塞的錐形瓶中,加入70%甲醇50 mL,蓋緊瓶塞,準確稱定總質量,超聲處理(功率250 W,頻率50 kHz)30 min,放冷,再稱定質量,用上述溶劑補足減失的質量,搖勻,濾過,取續濾液,即得供試品溶液[8-9]。同法制得缺赤芍、牡丹皮的陰性對照品溶液。取芍藥苷對照品適量,精密稱定,加甲醇制成含芍藥苷0.12 g/L的溶液,搖勻,即得對照品溶液。
2.3.3 方法學考察[2]
專屬性試驗:按2.3.1項下色譜條件,分別精密量取對照品溶液、供試品溶液和陰性對照品溶液各10 μL,注入液相色譜儀,測定,記錄色譜圖(圖2)。結果供試品溶液色譜中,在與對照品溶液色譜相同位置上有相同時間保留峰,而陰性對照無干擾。芍藥苷保留時間約為20.8 min,測得理論板數按芍藥苷峰計為14 506。
線性關系考察:分別精密吸取對照品溶液(117.408μg/mL)1,2,4,6,8,10 μL,注入液相色譜儀,按擬訂色譜條件進樣測定,以峰面積積分值(Y)為縱坐標、芍藥苷進樣量(μg,X)為橫坐標繪制標準曲線,得回歸方程Y = 1 242.638 8 X -7.596 3,r=0.999 8(n=6)。結果表明,芍藥苷進樣量在 0.117 408 ~ 1.174 080 μg 范圍內與峰面積積分值線性關系良好。
精密度試驗:精密吸取同一對照品溶液(117.408μg/mL)5 μL,重復進樣 6次。結果峰面積積分值的 RSD為0.19%(n=6),表明儀器精密度良好。
重復性試驗:取同一批(批號為20150701)供試品6份,依法制備供試品溶液(定容至50 mL具塞錐形瓶中),按擬訂色譜條件進樣,以芍藥苷峰面積積分值計算含量。結果見表1,可見,方法重復性良好。

圖2 杜仲片高效液相色譜圖

表1 重復性試驗結果(n=6)
穩定性試驗:取同一供試品溶液,注入液相色譜儀,分別于 0,2,4,8,12,16 h 時依法測定峰面積[10]。結果分別為 838.239 38,839.581 48,837.495 97,839.503 11,840.758 06,840.421 88,RSD 為 0.15% (n =6),表明供試品溶液在16 h內穩定。
加樣回收試驗:精密量取已知含量(批號為20150701,含量為 6.48 mg /g)的樣品 0.25 g,置 25 mL具塞錐形瓶中,分別精密加入芍藥苷對照品適量,按擬訂方法制備供試品溶液并測定。結果見表2。
2.3.4 樣品含量測定
精密稱取3批杜仲片,按擬訂方法制備供試品溶液,依法測定芍藥苷含量。結果批號為 20150701,20150901,20151101的3批樣品中芍藥苷含量分別為6.48,6.37,6.57 mg /g,平均含量為 6.473 mg /g。
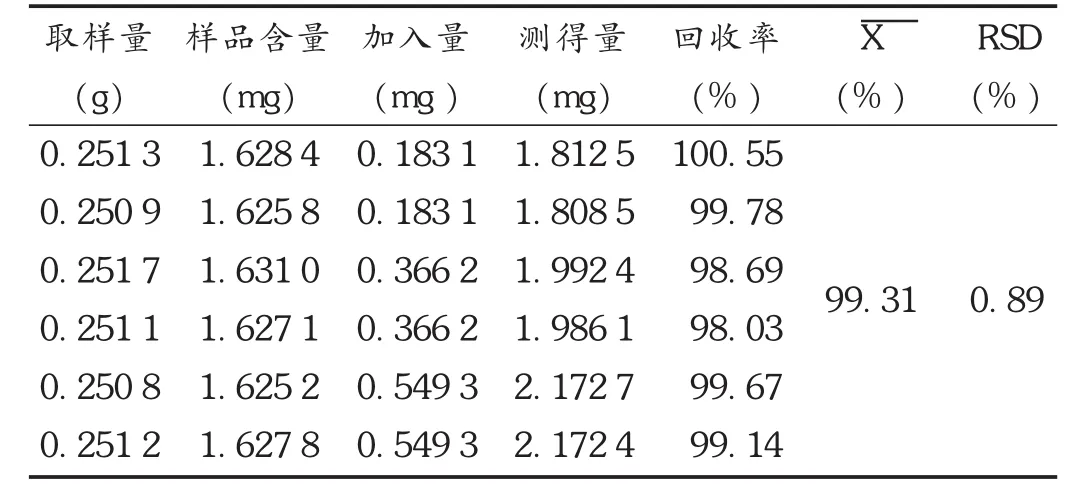
表2 芍藥苷加樣回收試驗結果(n=6)
3 討論
3.1 定性指標選擇
杜仲為杜仲片的君藥,松脂醇二葡萄糖苷為杜仲的主要有效成分,參考2015年版《中國藥典(一部)》的方法,色譜柱采用 Lichrospher-C18柱(250 mm ×4.6 mm,5 μm),流動相為乙腈 - 0.1% 磷酸溶液(13 ∶87),檢測波長為 227 nm,進樣量為10 μL,柱溫為30℃,對杜仲片中的松脂醇二葡萄糖苷進行測定,結果供試品溶液及陰性對照品溶液在與松脂醇二葡萄糖苷相同保留時間處均未檢出色譜峰,說明樣品中含量甚微,這可能與鹽杜仲的加熱炮制及杜仲片的生產工藝有關,故暫不建立杜仲片中松脂醇二葡萄糖苷的含量測定方法。
3.2 提取溶劑選擇
取杜仲片10片,搗碎成細粉,稱取約0.5 g,精密稱定,置具塞錐形瓶中,分別精密加入稀乙醇、70%甲醇、甲醇各50 mL,密塞,稱定質量,超聲處理(功率250 W,頻率50 kHz)30 min,放冷,再稱定質量,用上述溶劑補足減失的質量,搖勻,濾過,取續濾液,即得供試品溶液。結果用稀乙醇、70%甲醇、甲醇提取得芍藥苷含量分別為 4.02,6.57,5.16 mg /g。可見,采用 70% 甲醇作為提取溶劑,樣品中芍藥苷的含量明顯較高。
3.3 赤芍薄層鑒別
由于處方中赤芍飲片具有散瘀止痛的功效,故本研究中曾嘗試進行赤芍的薄層鑒別,分別采用多種提取方法(①正丁醇直接提取;②正丁醇直接提取后用氨試液洗滌;③正丁醇直接提取后用氨試液洗滌,取正丁醇層再用水洗滌等),并進行比較,最后選用背景干擾較少的方法③進行提取,陰性對照品溶液的制備方法同③。對提取液采用2種展開系統[氯仿-甲醇-醋酸乙酯(8∶3∶1)氨飽和 20 min,以及氯仿 -醋酸乙酯 -甲醇 -甲酸(40 ∶5 ∶10 ∶0.2)]進行展開鑒別,發現缺赤芍的陰性對照品溶液均有干擾,故不對此進行鑒別。
3.4 顯微鑒別
試驗中還曾嘗試進行杜仲的顯微鑒別,但由于投料加工后受其他藥材染色的影響,色較暗,與投料用的鹽杜仲在特征上雖有一定差異,但很難觀察。此外,杜仲藥材所具有的石細胞顯微特征,在制劑中并不具唯一性。因此,不對杜仲進行顯微鑒別。
[1]林向前 .杜仲片提取工藝研究[J].中國藥業,2013,22(13):32-33.
[2]邵紅燕,張和明,劉志海.骨質增生貼質量標準研究[J].中國藥房,2010,21(27):2563 - 2564.
[3]王麗聰,李 松.苦參配方顆粒質量控制研究[J].現代藥物與臨床,2012,27(5):471 - 473.
[4]國家藥典委員會.中華人民共和國藥典(四部)[M].北京:中國醫藥科技出版社,2015:3 -4,57-59.
[5]楊 艷,郝麗曉,李利芬,等.對中藥材市場丹參質量的考察[J].科技情報開發與經濟,2004,14(2):125 - 126.
[6]夏 荃.祛斑膠囊質量控制方法的研究[J].陜西中醫學院學報,2006,29(2):64 - 65.
[7]國家藥典委員會.中華人民共和國藥典(一部)[M].北京:中國醫藥科技出版社,2015:105.
[8]楊 培,李 建,徐 蓓,等.黔產山銀花栽培品種的鑒定與含量測定[J].中國藥業,2007,16(16):20 - 21.
[9]劉會前,李成網.十四味連黃燒傷軟膏的制備與質量控制[J].中國醫院藥學雜志,2009,29(16):1401 - 1403.
[10]李 虹.腎石通顆粒質量標準的研究[J].中國藥品標準,2010,11(3):176 - 179.
