新生兒第一二鰓弓綜合征一例報告
2019-04-09 10:16:28宋立劉洋王丹
天津醫藥 2019年3期
宋立,劉洋△,王丹
第一二鰓弓綜合征(first and second branchial arch syndrome),又稱為半側顏面短小畸形(hemifacial microsomia,HFM),是由于第一、二鰓弓發育不全導致的一種以顱面廣泛發育異常為特征的先天性畸形。HFM由Gorlin和Pindborg于1964年首先提出,并被歐美學者廣泛采用。HFM是新生兒中僅次于唇腭裂的第二大常見的先天性顱面出生缺陷[1]。目前病因不明,國外報道發病率約為活產兒的1/3 000~1/5 600[2],男女發病比例約為3∶2,右側顏面比左側更容易受累,比例接近3∶2[3]。在我國HFM病例偶有報道,但是仍缺乏系統的流行病學資料。雖然大多數HFM 病例是散發的,但大約2%~10%的病例是家族性的[4]。本文介紹1例HFM女嬰的臨床和影像學表現,以增強臨床醫生對這一少見病的認知和診斷能力。
1 病例報告
患兒 女,生后1個月。主因哭鬧時口角歪斜伴雙耳畸形,于2018年3月30日就診于我院。患兒系棄嬰,孕產史、家族史均不詳。入院查體:體質量4 160 g,身高53 cm,頭圍37 cm,胸圍36 cm,腹圍34 cm,神志清,精神反應可,呼吸平穩,無發紺,右側面部稍短小,右耳除耳垂正常外、耳廓上半部與顱側皮膚連成一片、牽拉耳垂可見狹窄外耳道、耳垂后上方可見一皮贅,左耳廓上部呈幕狀垂落、遮蓋對耳輪上角,哭鬧時口角向左下歪斜,伴右側鼻唇溝變淺,兩側眉毛高度、眼裂大小、閉眼及皺眉動作均未見異常,全身皮膚無皮疹、出血點、瘀斑及黃染,皮膚彈性好,前囟門張力不高,無頸亢,雙肺呼吸音清,未聞及干濕性啰音,心音有力,心律齊,腹軟,不脹,肝臟右肋下1.0 cm,質軟,四肢活動可,肌張力正常,擁抱反射、握持反射、覓食反射、吸吮反射均正常,手足末梢溫暖,脈搏有力,前臂內側毛細血管再充盈時間2 s(圖1)。血氣分析、電解質未見異常。肝腎功能、心肌酶未見異常。血、便常規未見異常。尿常規:鏡檢白細胞10~20 個/HP。血、尿培養陰性。肝炎病毒全項、梅毒、艾滋病抗體均陰性。巨細胞、弓形蟲、風疹病毒、單純皰疹病毒檢測均陰性。免疫球蛋白檢測未見異常。血尿代謝病篩查未見明顯異常。用CytoScan HD 芯片行染色體微陣列分析結果:arr(1-22,X)×2,未檢測到基因拷貝數缺失、重復和大片段純合子現象(圖2)。CT示腦外間隙增寬,右側耳廓形態較小,骨性外耳道狹窄,聽小骨形態不規則,中耳鼓室少量滲出,乳突小房氣化不良,篩竇黏膜增厚(圖3)。面神經肌電圖:右側面神經損害。腦干聽覺通路檢測:右耳于120 dB SL 可引出明確V 波,于102 dB SL 及110 dB SL 均未引出明確波形,右側聽力重度下降;左耳Ⅰ、Ⅲ、Ⅴ波可引出,左側各波潛伏期延長,間期正常,左側聽力中度下降。X線檢查示心、肺、膈及腹部未見異常,椎體未見異常(圖4)。B超:肝脾腎腦未見異常。心臟超聲:卵圓孔未閉(3 mm)。眼底檢查未見異常。診斷:(1)新生兒第一二鰓弓綜合征。(2)新生兒泌尿道感染。(3)卵圓孔未閉。患兒住院后給予拉氧頭孢鈉60 mg/(kg·d)靜脈滴注治療,外陰局部清潔護理,配方奶喂養。共住院13 d,復查尿常規未見異常,病情好轉出院。出院后分別于1個月、3個月、6個月定期隨訪,患兒體格發育正常,未復查聽力和CT。
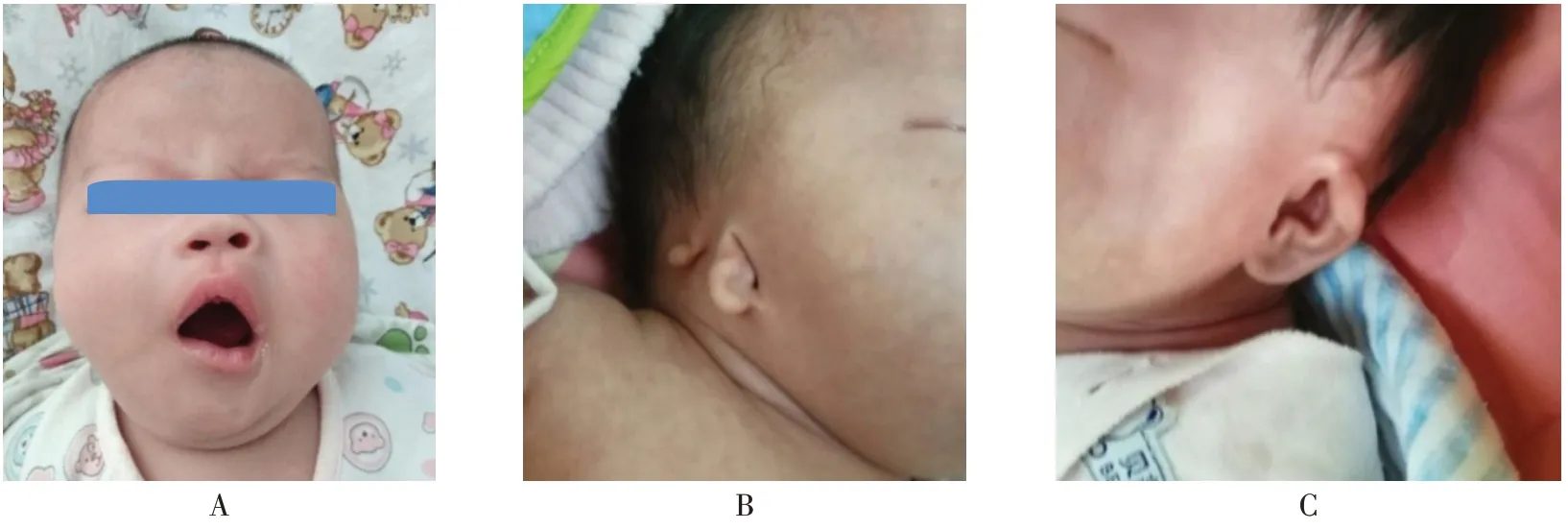
Fig.1 Clinical phenotype of the child with HFM圖1 HFM患兒臨床表型
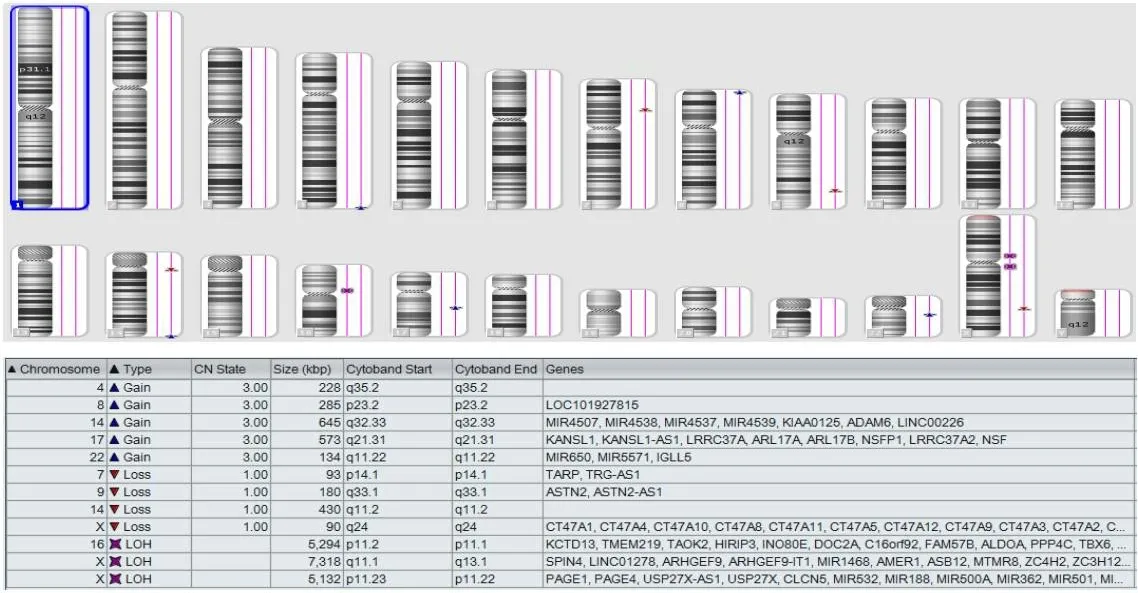
Fig.2 Chromosome microarray analysis of the child with HFM圖2 HFM患兒染色體微陣列結果
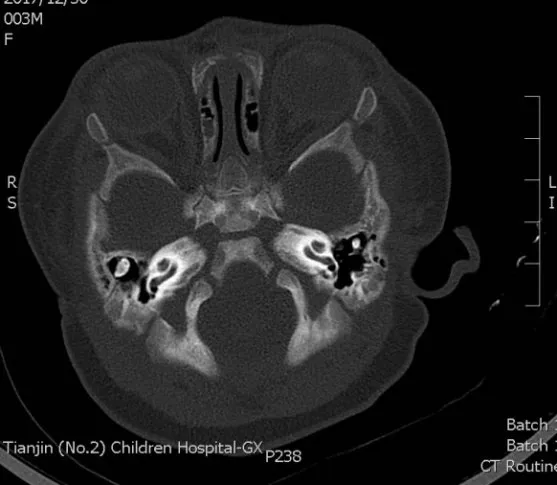
Fig.3 CT image of the child with HFM圖3 HFM患兒CT影像
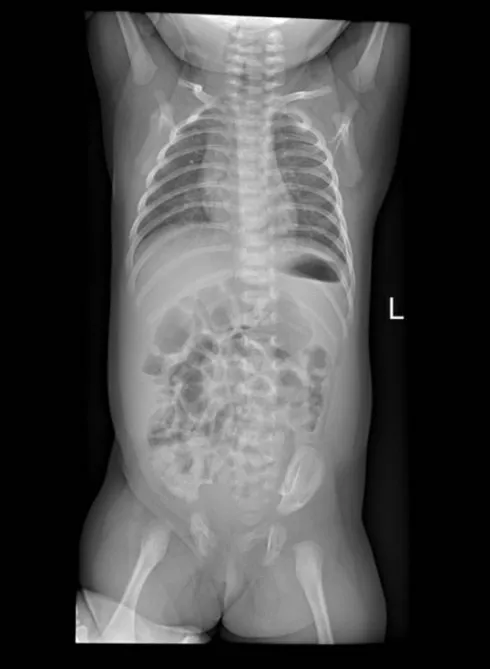
Fig.4 X-ray image of the child with HFM圖4 HFM患兒X線影像
2 討論
迄今為止,HFM 的病因及發病機制尚不清楚,已報道的各種致畸理論復雜,包括基因突變、子宮內血管破裂、染色體異常、第一和第二鰓弓畸形、生長激素失衡、血清素受體結合的變異、血管活性藥物的使用、缺氧、致畸胎素和妊娠期糖尿病等[5]。Zielinski等[4]對一個5代HFM大家系進行研究發現,OTX2可能是該家系HFM最有可能的致病基因。
HFM 有高度異質性,目前尚無統一的診斷標準[6]。輕度僅表現為單側小耳,可不伴有面部不對稱,嚴重者可累及頜骨、顳骨、顴骨、外耳和中耳的上升支等,表現為患側面部短小、耳閉鎖、眼眶不對稱、面橫裂、牙齒發育延遲等。隨著生長發育,患側面部骨骼生長落后,畸形逐漸加重。30%~50%的患者出現聽力減退[5],22%~50%的患者出現面癱[7]。影像學在HFM 診斷過程中起著重要的作用。CT 可以提供面部軟組織的三維再現和基礎骨的圖像,它有助于分析解剖畸形的程度和與相鄰顱面骨骼畸形的關系。而伴隨有椎體、心臟和眼部病變相關的HFM被稱為眼-耳-脊柱綜合征,也稱為Goldenhar 綜合征[8]。臨床上還應注意與歪嘴哭綜合征相鑒別,此病多有染色體22q11.2缺失,除表現為先天性單側下口唇麻痹外,還同時伴有多器官系統畸形。楊蕾等[9]認為,通過產前四維超聲檢查能夠直觀顯示胎兒的顏面部,在診斷HFM 和優生優育方面有重要價值。
本例患兒臨床主要表現為右側面部稍顯短小,右側耳廓畸形缺如伴外耳道狹窄,左耳耳廓上緣形態異常,哭鬧時口角向左下歪斜,CT 提示右耳廓形態較小、右側骨性外耳道狹窄、聽小骨形態不規則,頜骨未見明顯異常,面神經肌電圖提示右側面神經損害,診斷為HFM。患兒X 線片未見椎體病變,心臟超聲不支持先天性心臟病,眼底檢查無異常,不符合Goldenhar綜合征。染色體微陣列檢測無異常,不支持歪嘴哭綜合征等染色體異常相關疾患。由于本患兒系棄嬰,故不能完成基因相關研究,這也是本病例研究的不足之處。
HFM 的治療包括手術及非手術治療。重度HFM需要通過手術干預,比如牽張成骨術、耳再造、肋骨軟骨移植等[10]。本例患兒年齡小,骨骼肌肉軟組織發育尚未完全,需定期隨訪,觀察顱面部畸形進展,動態復查CT 了解頜骨等顏面骨骼發育,觀察牙齒生長情況,指導后期手術治療。隨著年齡及病情發展,HFM 患者會經歷視覺、聽覺、言語、進食和呼吸等不同程度的困難,對于負面情緒的心理疏導不容忽視。讓HFM患兒家長全面了解病情,穩定家長情緒并重拾信心,可以避免更多殘疾孩子因家長的絕望而被遺棄。
目前還沒有針對HFM 的嬰兒及兒童的大樣本研究,隨著遺傳學研究的進展,從分子生物學角度對于該病病因的研究會更加深入。而通過細致的體格檢查和影像學技術,可以為HFM 的診斷提供依據,手術及非手術治療已經有了很多的臨床成功經驗,同時對于HFM 家庭的心理社會支持也值得研究者更多關注。
