固相萃取GC-MS法測定生活用水中有機磷和擬除蟲菊酯類農藥殘留
2020-06-24 12:50:52嚴妍謝飛
綠色科技 2020年2期
關鍵詞:固相萃取
嚴妍 謝飛


摘要:采用全自動固相洗脫劑萃取GC- MS聯用法建立了對生活飲用水中常見的8種有機磷和擬除蟲菊酯類多種農藥化合物殘留的最佳化檢測方法。在對樣品中洗脫劑的種類與體積、洗脫劑量與流速、儀器的檢測條件等基礎上進行了最佳化,并在最佳化的條件下對洗脫劑進行了一系列的實驗回收檢測。結果表明:在優化的條件下,以上8種有機磷農藥在0.01-1.00 μg/mL濃度的范圍內呈現良好的線性相關性,相關系數0. 9922>0. 9913;不同種類和濃度的洗脫劑加標實驗回收加標實驗結果顯示其檢測方法的回收率基本穩定在90.4%~105.7%之間。加標實驗回收檢測結果也表明,此檢測方法具有樣品前處理中洗脫劑消耗有機溶劑量少、操作便捷、結果準確、靈敏度高等優點,能夠廣泛應用于對生活用水中多種常見的有機磷和擬除蟲菊酯類多種農藥化合物殘留的實驗檢測。
關鍵詞:固相萃取;GC- MS;農藥殘留
中圖分類號:X832 文獻標識碼:A 文章編號:1674-9944(2020) 2-009803
1 引言
農藥廣泛應用于我國現代農業的化學生產中,目前常見的農藥化學種類主要包括有機氯、有機磷和金屬擬除蟲菊磷酸酯類等幾類,其中有機氯類農藥因其化學毒性較大,農藥的殘留不易被人體自然吸收和降解,容易在復雜的生物鏈中持續產生長期積累的效應而產生對于生態環境持久嚴重的損害[1],因此逐漸被生產企業淘汰,而濫用有機磷和擬除蟲菊酯類的農藥盡管對環境的危害性一般相對有機氯農藥來說要低些,但是過度使用仍可對于人畜健康和生態環境帶來較大的危害,且此類有機化合物對于生活中的飲用水也往往具有較大的污染風險。由于有機磷類農藥化合物殘留在水中的含量往往遠遠低于儀器檢出限,因此需要采取有機溶劑濃縮富集等的預處理,提高其檢測的準確性[2,3]。目前,農藥化合物在檢驗前進行預處理的方法主要是液液的萃取法,由于液液的萃取法容易對環境產生二次污染,而且從農藥萃取到選樣工作的流程較為復雜,需要大量的技術人力和物力。固相化合物萃取技術可以將農藥的萃取、濃縮、選樣等多種功能技術匯集于一體,從而大幅度地優化和提升萃取工作效率,降低乃至于杜絕了可能在樣品中出現的人為技術操作失誤,因此,固相萃取質譜聯用技術已經成為了一種具有良好的應用和發展前景的對樣品前期分析處理的技術和方法[4,5]。而且還采用了氣相色譜一質譜聯用分析技術對樣品進行較高的穩定性和定量分析。該質譜聯用方法較之單純的氣相色譜法,具有溶劑用量少、回收率高、準確性好等優點,可有效廣泛運用于對生活用水中有機磷和擬除蟲菊酯類化合物和農藥的分析檢測。
2 實驗部分
氣相色譜一質譜聯用儀( GC- MS):Thermo Scien-tific IS0 7000單四級桿,美國賽默飛公司。
萃取裝置:Dionex 280固相萃取儀,美國賽默飛公司。
C18硅膠固相萃取小柱(40 m×0.3 mm,0.3μm,美國賽默飛公司)。
純水機:ULUP超純水機,優普儀器設備公司。
有機磷標準溶液(其中包含甲基敵敵畏、樂果、馬拉硫磷、對硫磷、甲基對硫磷5種),擬除蟲聚酯標準溶液(乙胺菊酯、甲氰菊酯、氯菊酯3種),以上各標準溶液濃度均為100 mg/L。
二氯甲烷、乙酸乙酯、甲醇鹽酸、丙酮、氫氧化鈉等試劑,均為光譜純。
(1)GC條件。毛細管固相萃取柱:C18硅膠固相萃取小柱(40 m×0.3 mm,0.3μm);載氣:99. 999%的高純氦氣,流速為6.0 mL/min。
儀器升溫程序如下所示:①50℃下持續1 min;②15℃/min升溫至170℃,恒溫3 min;③15℃/min升溫至230℃,恒溫3 min;④20℃/min升溫至280℃。
(2)MS條件。進樣溫度:260℃;離子源EI溫度:230℃;MS傳輸線溫度:280℃;四級桿溫度:150℃;溶劑延遲:3 min。
(3)水樣采集。棕色玻璃瓶裝滿采集水樣,4℃低溫保存,盡快分析水樣進行檢測。
(4)固相萃取。采用4.0 mL乙酸乙酯和二氯甲烷(1:1)的混和溶液,6.O mL的二氯甲醇及10.0 mL的鹽酸純水溶液活化固相萃取柱,流速平均為10.0 mL/min。取待測水樣1000 mL加入1L的玻璃瓶中,用6mol/L的鹽酸調節pH值在1.5~2之間,加入5 mL的甲醇,混勻后以20 mL/min的流速通過固相萃取柱。全部水樣萃取完成后,將固相萃取柱用氬氣吹干。并先后加入5.O mL乙酸乙酯和5.O mL二氯甲烷洗脫固相萃取柱,在測試管中合并上述洗脫液,氬氣吹至近干后,滴加乙酸乙酯溶液定容至2.O mL,上機待測。
(5)定量檢測。采用GC- MS在全掃描(scan)的模式下,以縮短標準樣品的保留使用時間和數據庫譜檢索主要進行化合物的定性離子分析;主要選擇放射性離子作為監測的模式,以各種類化合物的特定荷質和離子豐度進行定量分析,選擇特定的放射性離子時,應該首先選擇豐度含量比較高,荷質比較大的放射性離子,然后做為定量離子進行定量分析。
3 結果和討論
3.1 洗脫劑的選擇
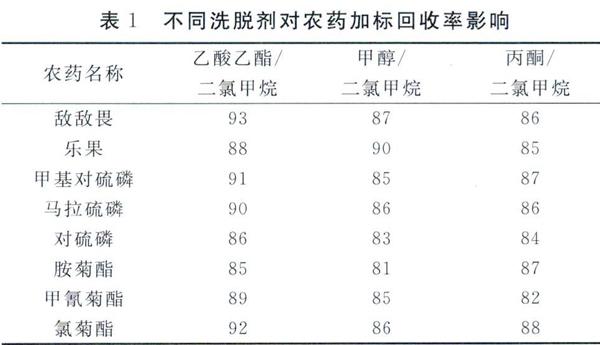
根據農藥洗脫劑的化學性質及c18萃取柱的化學穩定性及其特點,選擇常用的有機溶劑例如丙酮、甲醇、乙酸乙酯、二氯甲烷等,實驗中選擇兩種不同組分的混合溶劑作為洗脫溶劑,兩種組分濃度配比均為1:1,由于乙酸乙酯和二氯甲烷的化學極性較大,且二氯甲烷是固相萃取儀的常用萃取試劑,故在實驗中以乙酸乙酯和二氯甲烷的成份作為必加溶劑,選擇了3種可以混合的溶劑:乙酸乙酯十二氯甲烷、甲醇十二氯甲烷、丙酮十二氯甲烷,尋找最佳的洗脫濃度和溶劑的組合。本次實驗中選取洗脫濃度為0. 10μg/mL 8種混合農藥的回收率和混合溶液用磷做洗脫劑回收率穩定性實驗,結果統計見表1,根據8種混合農藥的溶劑回收率和洗脫劑回收穩定性的分析,用洗脫劑乙酸乙酯十二氯甲烷的混合溶劑效果比較好。
3.2 色譜條件最佳化
為了更好地尋找最優進行實驗的條件,在實驗過程中,對實驗程序中的樣品升溫、進入采樣口溫度、載氣流速及混合延遲時間分別進行了優化,得出上述的最佳進行實驗的條件,并在最佳實驗條件下,對樣品濃度為0.10 μg/mL的8種農藥的混合溶液上機進行測試,結果表明上述8種農藥在25 min內就可以得到良好的抗氧化和分離,保留時間、主要的定量離子和次要定性離子分別見表2所示。
在最佳條件下,以3倍信噪比所示的濃度為該方法的檢出限,得出8種農藥殘留的檢出限和測定下限如表3所示。
3.3 加標回收率實驗
以測定濃度0. 05、0.10、0.50 μg/mL的固相提取混合樣品濃度作加標回收率,每一個農藥混合樣品至少重復進行測定6次,測定的結果參見表4。
4 實驗結論
利用固相化合物萃取- GC - MS聯用可快速、準確地檢測出在生活用水中的常見有機磷和其他擬除蟲菊酯類農藥的有機磷殘留。通過一系列對條件比較優化的實驗,表明該檢測方法所需的有機磷溶劑量少、前處理方便、檢出限低、準確性好,加標有機磷回收率在90.4%~105.7%之間。這說明此檢測方法目前能有效地應用于對生活用水中常見的有機磷和擬除蟲菊酯類農藥的殘留的分析和檢測中,并達到要求。
參考文獻:
[1]吳自清,吳瓊,張青,等.固相萃取一氣質聯用法測定水中5種有機磷農藥[J].當代化工,2018,47(1):209-216.
[2]胡堪東,趙鳳英,萬益群.水中有機磷及氨基甲酸酯類農藥殘留量的GC - MS測定[J].南昌大學學報(理科版),2007,31(5):460-462.
[3]陳繼峰,林玉娜,李曉晶.氣相色譜串聯質譜法測定飲用水中多種農藥及其代謝產物殘留[Jl.醫學動物防制,2018,34 (5): 468-471.
[4]劉永波,賈立華,薛瑞芳,等.GC - MS法測定擬除蟲菊酯類農藥殘留[J].理化檢驗 化學分冊,2006,42(8):637-639,643.
[5]袁曉梅.氣相色譜一質譜聯用法測定水中有機磷農藥的研究[J].環境與發展,2019(5):176-177.
作者簡介:嚴妍(1994-),女,助理工程師,研究方向為土壤與水質分析檢測。
猜你喜歡
分析化學(2016年7期)2016-12-08 00:54:07
中國科技博覽(2016年2期)2016-04-25 14:11:43
湖北工業職業技術學院學報(2016年1期)2016-04-20 17:12:54
分析化學(2015年10期)2015-11-03 07:52:24
食品安全導刊(2015年10期)2015-10-26 04:44:22
安徽農學通報(2015年18期)2015-10-20 00:50:11
安徽農學通報(2015年17期)2015-09-30 00:52:24
分析化學(2015年9期)2015-09-11 07:09:54
肉類研究(2015年5期)2015-08-08 12:46:08
肉類研究(2015年3期)2015-06-16 12:40:36
