氣相色譜法測定水果中咪鮮胺殘留量的前處理方法探究
2020-07-18 04:06:46劉志鵬楊李勝黃敏興柯華南
食品工業(yè) 2020年6期
關鍵詞:方法
劉志鵬,楊李勝*,黃敏興,柯華南
1. 廣東省生物工程研究所/廣州甘蔗糖業(yè)研究所(廣州 510000);2. 中國輕工業(yè)甘蔗制糖工程技術研究中心(廣州 510000)
咪鮮胺又名施保克,其通用名為Prochloraz,化學名為N-丙基N-[2-(2, 4, 6-三氯苯氧基)乙基]-1咪唑基甲酰胺(LPAC),在動植物體內的最終代謝產物為2, 4, 6-trichbrophenbor(2, 4, 6-TCP)[1]。咪鮮胺及其制劑進入我國市場后,推廣應用發(fā)展很快,已經被廣泛應用于多種農林產品的生產、貯存和運輸等過程中[2]。咪鮮胺是一種咪唑類廣譜殺菌劑,可用于谷類、熱帶和亞熱帶水果、蔬菜等經濟作物的病蟲害的防治[3]。盡管咪鮮胺是低毒殺菌劑,但其最終產物2,4, 6-三氯苯酚是環(huán)境中的重要污染物之一,會污染生態(tài)環(huán)境,并會對人類健康造成潛在威脅。因此,其在水果生產和儲藏中的殘留問題越來越受到人們關注[4]。咪鮮胺最大殘留限量被美國和歐盟嚴格監(jiān)測,在水果中的最大殘留限量(MRL)一直在很低的水平上[5]。
目前,國內外已發(fā)表的對咪鮮胺殘留物的檢測主要采用色譜法[6],主要是氣相色譜-質譜聯(lián)用[7]、高效液相色譜-質譜、高效液相色譜和氣相色譜。尹桂豪等[8]采用氣相色譜法對芒果中咪鮮胺殘留分析;韓麗君等[9]采用GC-ECD研究了咪鮮胺及其代謝物在水稻中的總殘留量檢測方法;De-Paoli等[10]對咪鮮胺及其代謝產物殘留在蔬菜、水果和小麥樣品組合簡化測定氣相色譜法。
但這些方法卻存在著一些問題:一是氣相色譜-質譜聯(lián)用和高效液相色譜-質譜的前處理方法雖然簡單,但儀器昂貴且難以普及應用;二是使用高效液相色譜只可以檢測咪鮮胺的殘留量,但不能檢測其全部的代謝物,尤其是2, 4, 6-三氯苯酚的殘留量,且靈敏度比氣相色譜低[4];三是樣品前處理方法技術落后,在處理大量樣品時需要消耗大量的試劑、玻璃儀器和時間,效率較低。
此次試驗同樣采用氣相色譜法對水果中的咪鮮胺及其代謝產物殘留檢測方法進行研究,對樣品前處理方法進行優(yōu)化,縮短提取過程的時間也能最大限度地提取咪鮮胺及其代謝產物;減少多余的玻璃儀器使用;并且采用一種新的方法進行凈化,避免了使用濃硫酸帶來的安全性隱患以及在操作過程中調節(jié)pH帶來的不確定性,提高了方法的選擇性和靈敏度,從而建立一套快速、便捷、安全、清潔、耗時短、低成本且靈敏度高的檢測方法。
1 材料與方法
1.1 材料與儀器
試樣蘋果、葡萄、西瓜、柑橘等水果,市售。
咪鮮胺、2, 4, 6-三氯苯酚(純度均不少于99.0%),德國DR公司;吡啶鹽酸鹽、鹽酸、濃硫酸、無水硫酸鈉(使用前在55 ℃下烘干2 h,冷卻后使用),均為分析純,廣州化學試劑廠;二氯甲烷、丙酮、石油醚、乙腈、正己烷,均為色譜級,美國默克公司;試驗用水均為一級水。
7890B氣相色譜儀,美國安捷倫公司;TLE204E電子天平,梅特勒-托利多儀器(上海)有限公司;DK-2可調式電砂浴鍋,北京市永光明醫(yī)療儀器有限公司;RV 8V旋轉蒸發(fā)儀,艾卡(廣州)儀器設備有限公司(德國IKA);SYG-1230水浴箱,上海珂淮儀器有限公司;T25均質機,艾卡(廣州)儀器設備有限公司(德國IKA)。
1.2 方法
1.2.1 標準溶液配制
咪鮮胺標準工作液:稱取10 mg咪鮮胺標準物質,用丙酮溶解并定容至100 mL,配制成質量濃度為100 mg/L的標準溶液儲備液,用丙酮再次稀釋配制成質量濃度為1.0 mg/L的咪鮮胺標準工作液。
2, 4, 6-三氯苯酚工作液:稱取10 mg 2, 4, 6-三氯苯酚標準物質,用丙酮溶解并定容至100 mL,配制成質量濃度為100 mg/L的標準溶液儲備液,用丙酮再次稀釋配制成質量濃度為1.0 mg/L的2, 4, 6-三氯苯酚標準工作液。
1.2.2 水果儲備基質液
分別稱取5份40 g蘋果試樣于5個100 mL的平底燒杯中,各加入50 mL的乙腈,勻漿機高速勻漿2 min,過濾,收集于裝有5 g氯化鈉的100 mL具塞比色管中,蓋上蓋子,劇烈振蕩1 min,靜置30 min,從比色管中吸取上層溶液于250 mL的平底燒杯中,搖勻,備用。
1.2.3 GC色譜條件
檢測方法采用GC-ECD法,色譜柱采用DB-5(30 m×0.320 mm×0.25 μm)的彈性石英毛細管柱。儀器檢測條件:柱溫70 ℃以40 ℃/min的速度升溫到245℃(保留5 min);進樣口溫度260 ℃;ECD溫度300℃;分流進樣,分流比40∶1;色譜柱的載氣氣體為氮氣,流量2.5 mL/min;進樣量1.0 μL。
1.2.4 標準曲線制作
準確吸取0.25,0.50,1.0,2.0和3.0 mL咪鮮胺標準工作液于水解試管中,氮吹濃縮近干,加入5 g吡啶鹽酸鹽,水解方法、凈化方法按照最優(yōu)方法進行,待測。
2 結果與分析
2.1 對前處理凈化方式優(yōu)化分析
分3組,每組各取6個50 mL平底燒杯并分別加入10 mL儲備基質液,然后在燒杯中分別加入1.0 mL 1.0 mg/L的2, 4, 6-三氯苯酚工作液(模擬咪鮮胺已完全水解),在振蕩器上混勻,放在氮吹儀上濃縮近干,待用。
1) 硫酸的凈化方法:將上述燒杯用20 mL正己烷分數次沖洗并全部轉移到分液漏斗中,加入5 mL硫酸,振蕩1 min,靜止分層后,棄去硫酸,重復3次。然后用蒸餾水洗滌有機相中的殘留硫酸,洗滌至中性,收集有機相,經無水硫酸鈉脫水干燥后濃縮低于5 mL,并用正己烷定容至5 mL,上機測定,結果如圖1所示。

圖1 利用濃硫酸進行凈化的咪鮮胺殘留量色譜圖
2) PSA 100 mg+C18100 mg+MgSO4300 mg QuECh-ERS凈化管的凈化方法:將上述燒杯用正己烷分數次洗滌燒杯并全部轉移到5 mL容量瓶,并定容至5 mL。然后全部轉移到裝有PSA 100 mg+C18100 mg+MgSO4300 mg的QuEChERS凈化管中凈化,上機測定,結果如圖2所示。
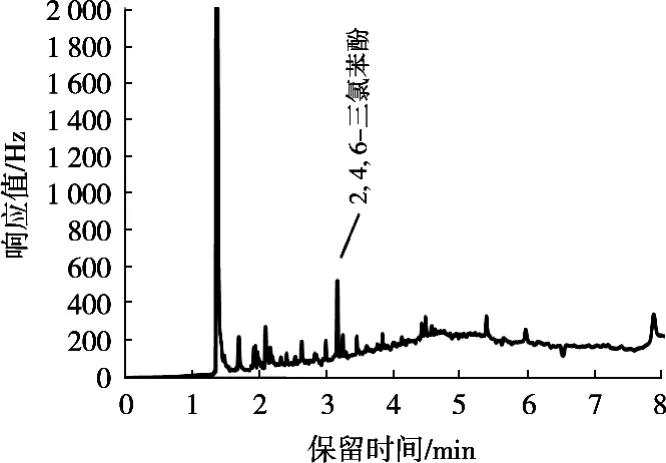
圖2 利用凈化管進行凈化的咪鮮胺殘留量色譜圖
3) 弗羅里硅土柱的凈化方法:將上述燒杯分數次加入5 mL正己烷,并全部轉移到弗羅里硅土柱上(已用洗脫液活化),棄去濾液,用25 mL丙酮-正己烷(90∶10,V/V)洗脫溶劑洗脫,用25 mL帶刻度試管接收濾液,氮吹濃縮低于5 mL,最后用正己烷定容至5 mL,上機測定,結果如圖3所示。

圖3 用弗羅里硅土柱進行凈化的咪鮮胺殘留量色譜圖
經計算,2, 4, 6-三氯苯酚回收率得到的結果如表1所示。由圖譜和回收率計算結果可知,QuEChERS凈化管的填料對樣品中2, 4, 6-三氯苯酚的吸附作用很強,能把試樣中大部分2, 4, 6-三氯苯酚吸附在凈化管中,并且對于其他雜峰的凈化效果不顯著,甚至是沒有得到凈化效果,影響咪鮮胺最后的定量計算,故排除使用QuEChERS凈化管的使用;而濃硫酸和弗羅里硅土柱兩種的凈化效果能達到檢測要求,回收率均在90%以上,但凈化過程使用濃硫酸,容易發(fā)生危險,而弗羅里硅土柱凈化則使用過程相對簡單,不會對實驗人員造成潛在的危險以及在之后調節(jié)pH的不確定性也可以避免。故使用弗羅里硅土柱作為凈化的裝置。
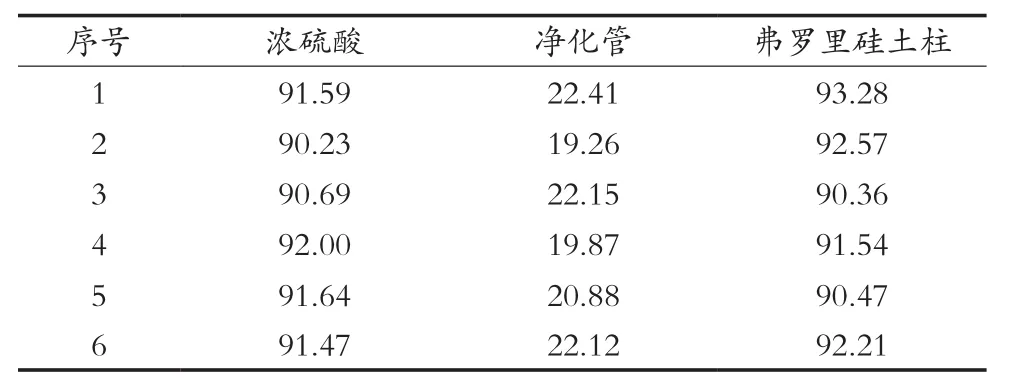
表1 不同凈化方法下的2, 4, 6-三氯苯酚回收率 %
2.2 對洗脫溶劑優(yōu)化分析
分3組,每組各取6個50 mL平底燒杯并分別加入10 mL儲備基質液,然后在燒杯中分別加入1.0 mL 1.0 mg/L的2, 4, 6-三氯苯酚工作液(模擬咪鮮胺已完全水解),在振蕩器上混勻,放在氮吹儀上濃縮近干,加入5 mL正己烷,蓋上蠟膜,待用。
根據2.1優(yōu)化,將弗羅里硅土柱用5 mL表2中不同配比洗脫液進行條件活化,當溶劑液面到達柱吸附層表面時,馬上倒入上述樣品溶液,用25 mL的具刻度試管收集洗脫液,分別用25 mL相同配比的溶液沖洗燒杯后洗脫凈化柱。將裝有洗脫液的試管置于氮吹儀,在50 ℃水浴中濃縮近干,用正己烷準確定容至5 mL,混勻,待測,結果如表2所示。
結果顯示,在丙酮與正己烷的配比是95∶5(V/V)和90∶10(V/V)及丙酮與二氯甲烷的配比是95∶5(V/V)時,2, 4, 6-三氯苯酚幾乎能全部被洗脫下來,其回收率均在90%以上,均滿足對檢測的要求。但是95∶5(V/V)的丙酮+二氯甲烷同時會對雜質的回收率也明顯增加,特別對咪鮮胺臨近雜峰的洗脫,此雜峰會影響咪鮮胺的的定量檢測,而在95∶5(V/V)和90∶10(V/V)的丙酮+正己烷洗脫溶劑中,由于丙酮價格比正己烷的高,為降低檢測成本,選擇90∶10(V/V)的丙酮+正己烷相對合適。
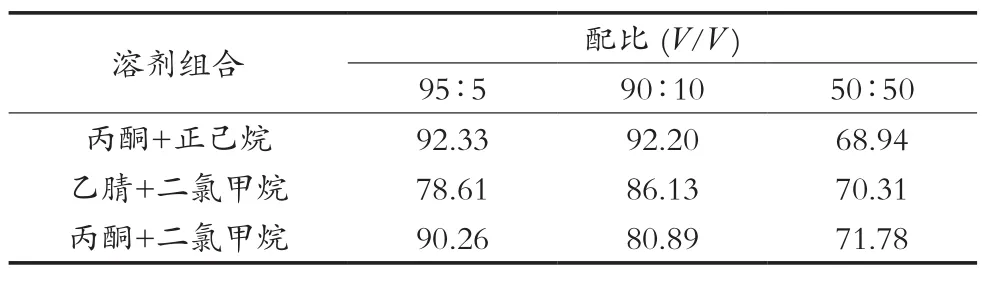
表2 不同洗脫溶劑的配比下2, 4, 6-三氯苯酚平均回收率 %
2.3 對水解裝置優(yōu)化分析
分3組,每組各取6支帶具塞刻度的25 mL水解試管,加入1.0 mL咪鮮胺標準工作液,氮吹濃縮近干,加入5 g吡啶鹽酸鹽,按照表3中的水解條件進行水解,冷卻至室溫后,加入適量的水溶解吡啶鹽酸鹽,并全部轉移到250 mL的分液漏斗中,試管用50 mL水分數次沖洗并轉移到分液漏斗中,石油醚萃取2次(每次50 mL),棄去水相,合并有機相;濃縮近干,凈化方法按照2.1、2.2優(yōu)化出的最優(yōu)方法進行。
經多次反復試驗,在沙浴與25 mL的水解試管裝置下,一個砂浴鍋能同時加熱20~30個試樣,同時進行水解,完全可以進行大批量處理樣品。水解過程并沒有沸騰現(xiàn)象,安全可靠。同時,反應后的試管容易清洗,能大大增加工作效率。其余方式操作繁瑣,不利于檢測多個樣品,不能提高處理效率。

表3 水解裝置及條件表
2.4 對提取溶液優(yōu)化分析
分3組,分別稱取40 g蘋果試樣于250 mL的平底燒杯中,加入1 mL 1.0 mg/L的咪鮮胺標準工作液,按照表5設計對應加入各種用量的溶劑,并在勻漿機上高速勻漿2 min,過濾,收集濾液于裝有5 g氯化鈉的100 mL具塞試管中,蓋上蓋子,劇烈振蕩1 min,在室溫下靜置30 min,使之分層,吸取20 mL上層溶液于25 mL的帶刻度的試管中,氮吹儀上濃縮近干;加入5 g吡啶鹽酸鹽,水解方法使用按照2.3篩選出的最佳水解裝置進行水解,水解1.5 h;凈化方法按照2.1、2.2優(yōu)化出的最優(yōu)方法進行,上機待測,咪鮮胺回收率結果見表4。
NY/T 1456—2007中前處理提取液是用80 mL丙酮,并且需要浸泡過夜后仍需要振蕩60 min,試驗時間長,不便于檢測短時高效的要求。上述結果表明丙酮和乙腈在適當的體積中均能滿足檢測要求,而乙腈的提取回收率均在90%以上,溶劑用量各水平差異不明顯。故使用乙腈作為提取溶劑,其用量為50 mL。
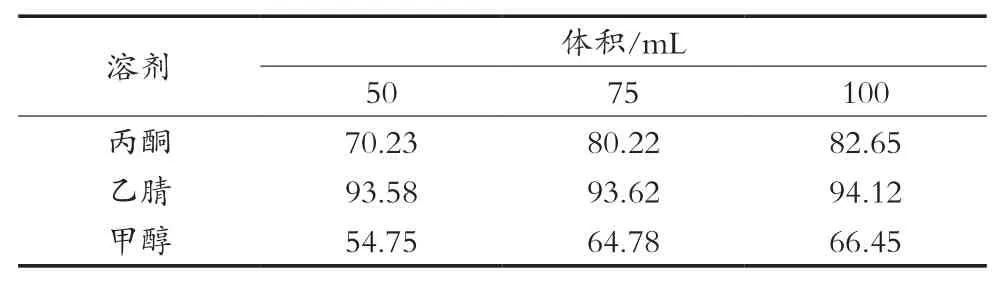
表4 不同提取溶劑試驗下咪鮮胺平均回收率 %
2.5 方法的線性范圍和檢出限
將1.2.4配制好的咪鮮胺標準曲線按照上述優(yōu)化后的條件進行處理進樣,以質量濃度(mg/L)為橫坐標,峰面積為縱坐標,繪制標準曲線(見圖4)。線性方程為y=12 991.72x-118.34,其相關性r=0.999 53,這說明在0.05~0.6 mg/L線性范圍內有良好的線性關系。通過實際低濃度添加試驗,當信噪比等于3時,咪鮮胺的檢出限為0.005 mg/kg,滿足試驗要求。

圖4 咪鮮胺標準工作曲線
2.6 方法的回收率和精密度
按照上述優(yōu)化后的條件,對蘋果、葡萄、西瓜、柑橘4種樣品分別添加0.05,0.20和0.40 mg/kg三個濃度水平,做加標回收試驗,每個濃度6平行,結果見表5。咪鮮胺的平均回收為87.2%~101.1%,相對標準偏差為2.1%~5.5%。
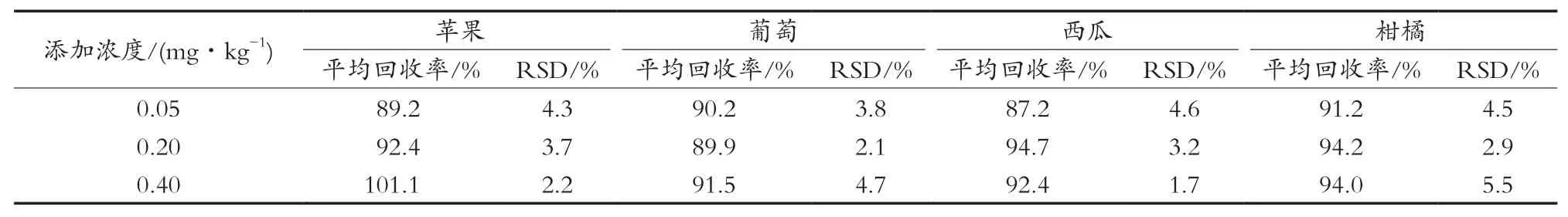
表5 4類蔬果在3個添加水平下的平均回收率和相對標準偏差(n=6)
2.7 方法在實際樣品上的應用
在廣州市各大市場上隨機抽取橙子、桃子、草莓、葡萄、西瓜、蘋果等水果,共109批次,按照優(yōu)化后的試驗條件,每批次3平行進行檢測。結果顯示共21批次檢出咪鮮胺及其代謝物,其中葡萄與柑橘類檢出率最高。其濃度范圍在0.011 1~0.291 mg/kg之間,RSD范圍在0.9%~7.2%之間。

圖5 葡萄陽性樣品色譜圖
圖6 0.2 mg/L 2, 4, 6-三氯苯酚標準點色譜圖
3 結論
此次試驗建立在GC-ECD檢測器的基礎上。經試驗驗證,測定水果中咪鮮胺殘留量的前處理方法優(yōu)化為:稱取40 g試樣于250 mL燒杯中,加入50 mL乙腈,高速均質2 min,過濾;收集濾液于裝有5 g氯化鈉的100 mL具塞試管中,蓋上蓋子,劇烈振蕩1 min,在室溫下靜置30 min,使之分層,吸取20 mL上層溶液于25 mL的帶刻度的水解試管中,氮吹濃縮近干后加入5 g吡啶鹽酸鹽,置于210~240 ℃的沙浴中水解1.5 h;冷卻后,加入10 mL蒸餾水使吡啶鹽酸鹽溶解,并用50 mL蒸餾水分次沖洗水解管轉移到250 mL分液漏斗中,用石油醚萃取2次(每次50 mL),合并有機相;將有機相轉移到100 mL燒杯中,濃縮至干,用10 mL正己烷分次洗燒杯,溶液轉移到經丙酮-正己烷混合溶液(90∶10,V/V)活化的弗羅里硅土柱,棄去濾液,用25 mL丙酮-正己烷混合溶液(90∶10,V/V)分數次洗脫,收集濾液,氮吹并定容5 mL,供測定。該前處理方法在保證符合檢測要求的基礎上,簡化了實驗步驟,優(yōu)化了實驗的條件,能同時檢測多個樣品,既縮短檢測的時間,又節(jié)省成本費用,安全可靠,方法簡單,且靈敏度高,適用于批量水果樣品中咪鮮胺及其代謝物殘留的定性和定量分析。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
