不同鈦源改性HZSM-5耦合低溫等離子體放電催化生物油提質反應的性能
2021-01-04 08:43:10樊永勝侯光喜熊永蓮蔡憶昔趙衛東
石油學報(石油加工) 2020年5期
樊永勝, 侯光喜, 熊永蓮, 蔡憶昔, 趙衛東
(1.鹽城工學院 汽車工程學院,江蘇 鹽城 224051;2.江蘇大學 汽車與交通工程學院,江蘇 鎮江 212013)
生物質熱解可獲得高附加值石油燃料替代品和基礎化工原料,受到眾多學者關注。目前,生物質催化熱解液化和液化生物油品質提升成為研究的熱點[1]。其中,HZSM-5分子篩因催化生物油提質效果明顯而得到廣泛關注[2]。生物油提質在HZSM-5催化下主要發生裂化和芳構化反應:生物油分子首先在L酸中心的作用下斷裂脫氧或脫氫生成碳正離子,然后在B酸中心上經過β位斷裂生成以C1~C4烯烴為主的碎片,然后繼續在B酸中心的作用下發生齊聚環化反應,最后在L酸中心上環脫氫生成芳香烴。然而,在生物油催化提質過程中,常規HZSM-5易結焦失活[3],且催化產物中多環芳香烴含量過高[4]而不能滿足要求,因此需要對HZSM-5進行改性以提升其性能。
迄今,眾多研究者對HZSM-5進行了化學改性,引入P、B等非金屬元素和Zn、Ni、Co、Ga等金屬元素[5-8],在一定程度上提高了催化劑的選擇性,增強了穩定性。然而,目前對HZSM-5進行Ti改性的研究仍然有限。Li等[9]對ZSM-5進行了Ti改性研究,發現Ti在催化過程中會發生價態變化,該過程中的電子遷移有利于促進碳正離子反應的進行;同時,Ti離子較高的價態和較小的半徑使其具有較強的極化能力,可明顯增強催化劑性能。在本課題組前期研究中,利用Ti/HZSM-5進行了生物油的提質研究,發現對HZSM-5進行Ti改性可明顯提高催化劑對輕質烴的選擇性,延緩催化劑結焦失活[10]。
低溫等離子體放電(Cold Plasma Discharge, CPD)協同催化是將高壓放電所引發的自由基反應與催化反應進行有機結合的一種新型催化處理技術。其中,高壓電場提供了大量能量,用于分解、激發和電離反應物原子和分子,使反應體系內富含電子、離子、自由基和激發態分子,改變了反應物在催化劑表面的化學吸附行為,提升了催化劑活性和化學反應的選擇性[11]。當HZSM-5催化劑催化反應過程中耦合CPD技術后,會在催化劑孔道及顆粒之間產生微放電,產生能量直接活化和解離反應物分子;同時,CPD技術產生的高能活性電子、離子等會撞擊催化劑表面形成等離子體鞘,減少催化劑結焦,提高催化效率。
在本課題組前期研究中,將CPD技術引入生物油催化裂解過程,設計了以介質阻擋放電為工作原理的生物油催化提質反應器,構建了基于CPD的在線催化提質系統[12]。研究表明:CPD的協同作用使催化劑有效工作溫度從550 ℃左右下降至400 ℃,且催化脫氧性能提升;在CPD作用下Ti改性HZSM-5對單環芳香烴和輕質脂肪烴的選擇性較高;然而精制生物油產率與燃料品位很難同時得到提升,Ti改性的作用機制及改性催化劑與CPD的耦合作用機制尚不明確;并且催化劑使用后結焦焦炭會出現“同構化”,即不同類型焦炭的熱分解規律趨于一致。
因此,筆者對HZSM-5進行不同Ti源改性,制備不同鈦改性催化劑,考察不同鈦改性催化劑耦合CPD對精制生物油產率、燃料品位及化學組成的影響。同時,為更好地分析鈦改性的作用機制,對反應過程中產生的高能活性自由基進行在線監測;并分析使用后催化劑的結焦特性,探究不同鈦改性對催化劑穩定性的影響及作用機制,為實現生物油高效提質提供依據。
1 實驗部分
1.1 原料
所用油菜籽殼采自江蘇省鹽城市某農場,將干油菜籽殼粉碎成粒徑為0.1~1.0 mm的顆粒,于105 ℃干燥24 h,保存備用。油菜籽殼的熱揮發組成、元素組成及高位熱值(HHV)如表1所示。一般而言,生物質或生物油的高位熱值越大,燃燒所能釋放的熱能就越多,燃料的品位相對越高。因此,生物油高位熱值的大小可以在一定程度上反映其燃料品位的高低。

表1 油菜籽殼的熱揮發組成、元素組成及高位熱值(HHV)Table 1 Thermal volatile composition, elemental composition and HHV of rapeseed shell
1.2 改性HZSM-5制備及表征
1.2.1 改性催化劑的制備
將硅/鋁原子比為50的HZSM-5原粉在550 ℃煅燒2 h后,將其分別浸漬到金紅石型TiO2和銳鈦型TiO2的水分散液(2種TiO2粉體粒徑約為1.6 μm),以及TiCl3和TiCl4的鹽酸溶液中,于80 ℃恒溫攪拌4 h,過濾、洗滌,于105 ℃干燥4 h以除去水分和鹽酸,最后于馬弗爐中550 ℃高溫焙燒4 h,得到Ti質量分數占HZSM-5的5%改性催化劑。分別將金紅石型TiO2、銳鈦型TiO2、TiCl3和TiCl4改性催化劑記為TiRH-5、TiAH-5、Ti3H-5和Ti4H-5。
1.2.2 改性催化劑的表征
采用德國布魯克AXS D8 Advance型X射線衍射(XRD)儀測定催化劑物相,以CuKα(λ=0.15406 nm)為輻射源,管電壓為40 kV、管電流為30 mA,掃描速率為5°/min,掃描范圍為2θ在5°~50°。
采用北京彼奧德SSA4300型分析儀進行液氮吸附-脫附測定催化劑比表面積和孔體積。由BET模型計算催化劑的比表面積;由BJH模型計算催化劑孔體積。
采用美國賽默飛Frontier型紅外光譜儀配合真空吡啶吸附-脫附系統測定催化劑的Br?nsted酸和Lewis酸分布。
1.3 生物油催化提質實驗系統和方法
1.3.1 實驗系統
生物油在線催化提質系統如圖1所示。該系統包括生物質熱解反應器、催化反應器、連接管路、過濾裝置、冷凝收集裝置、集氣裝置、穩壓筒、真空泵以及溫控裝置等。催化反應器采用介質阻擋放電(DBD)原理設計成同軸雙介質結構,具體結構參數見文獻[12],催化劑層置于放電區中。實驗時通過調壓閥調節體系內部壓力。此外,在催化反應器中填裝催化劑時,利用帶篩孔的石英玻璃薄片沿徑向隔開,形成一條細小的光學透光通道,用來實時檢測反應自由基中間體。
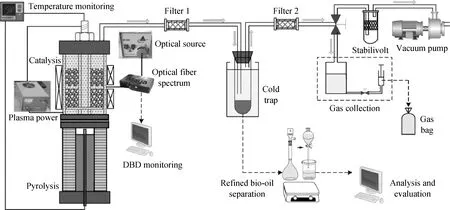
圖1 生物油在線催化提質系統Fig.1 Diagram of experimental apparatus of bio-oils online catalytic upgradingDBD—Dielectric barrier discharge
1.3.2 實驗方法
生物質先在反應器下段進行熱解,產生的熱解氣在真空泵抽吸作用下進入反應器上段進行催化提質反應,然后經過濾、冷凝等,得到液相生物油。用二氯甲烷萃取分離液相生物油中的有機物,得到精制生物油。實驗條件:催化劑層高度為30 mm;催化溫度為400 ℃;生物質熱解終溫為500 ℃;升溫速率為20 ℃/min;體系壓力為5 kPa;加載等離子體電源進行高壓高頻放電,輸出功率為800 W,輸出電流為30 mA。此條件下,熱解和催化過程均達到較優狀態[13]。
實驗過程中,采用美國海洋光學USB4000光纖光譜儀進行反應生成自由基分析,以美國海洋光學DH-2000為光源,檢測波段為350~1050 nm。
以生物質原料質量為基準,計算精制生物油產率。為便于分析,將HZSM-5、TiRH-5、TiAH-5、Ti3H-5和Ti4H-5催化得到的精制生物油分別記為 RB(0)、RB(R)、RB(A)、RB(3) 和RB(4)。對使用3次后的催化劑進行分析以評價其穩定性。
1.4 精制生物油及催化劑分析
1.4.1 精制生物油分析
采用意大利Euro Vector EA3000型元素分析儀測定精制生物油的元素組成。
采用美國賽默飛世爾Trace DSC II型氣-質聯用分析儀(GC/MS)測定精制生物油組成。GC條件:載氣為高純He,流量為1 mL/min,進樣口溫度為250 ℃,不分流,進樣量1 μL;MS條件:離子源溫度為230 ℃,MS傳輸線溫度為250 ℃,電離方式為EI,電子轟擊能量為70 eV,掃描質荷比范圍為30~500,掃描時間為1 s;升溫程序:30 ℃保持2 min,以15 ℃/min升溫至100 ℃,然后以 10 ℃/min 升至250 ℃并保持3 min,溶劑(CH2Cl2)延遲時間為3 min。
1.4.2 催化劑結焦分析
采用美國賽默飛世爾TGA/DSC 1型同步熱分析儀對結焦催化劑進行熱重分析。采用荷蘭飛利浦CM200型透射電子顯微鏡(TEM)觀察結焦催化劑上的焦炭分布及特征,點分辨率為0.235 nm。
2 結果與討論
2.1 改性催化劑表征分析
2.1.1 XRD分析
鈦改性前后催化劑X射線衍射譜如圖2所示。由圖2可見,各催化劑X射線衍射譜(JCPDS card:PDF 44-0003)基本一致,并未發生明顯的衍射峰位置偏移,表明鈦改性催化劑結構完整,未發生晶相取代。對于TiRH-5,在2θ為27.45°、54.32°和36.09°等位置有微弱的衍射峰,歸屬于金紅石型TiO2(JCPDS card:PDF 21-1276);對于TiAH-5,在2θ為25.28°、48.05°和37.80°等位置的衍射峰由銳鈦型TiO2(JCPDS card:PDF 21-1272)引起;對于Ti3H-5,由XRD圖可以確定HZSM-5負載TiCl3改性,催化劑上引入了多種中間價態的鈦氧化物,包括Ti2O3(JCPDS card:PDF 10-0063)和Ti3O5(JCPDS card:PDF 09-0309)等;對于Ti4H-5,XRD衍射峰分析表明,HZSM-5負載TiCl4改性,催化劑上引入的是常規TiO2((JCPDS card:PDF 65-5714)。
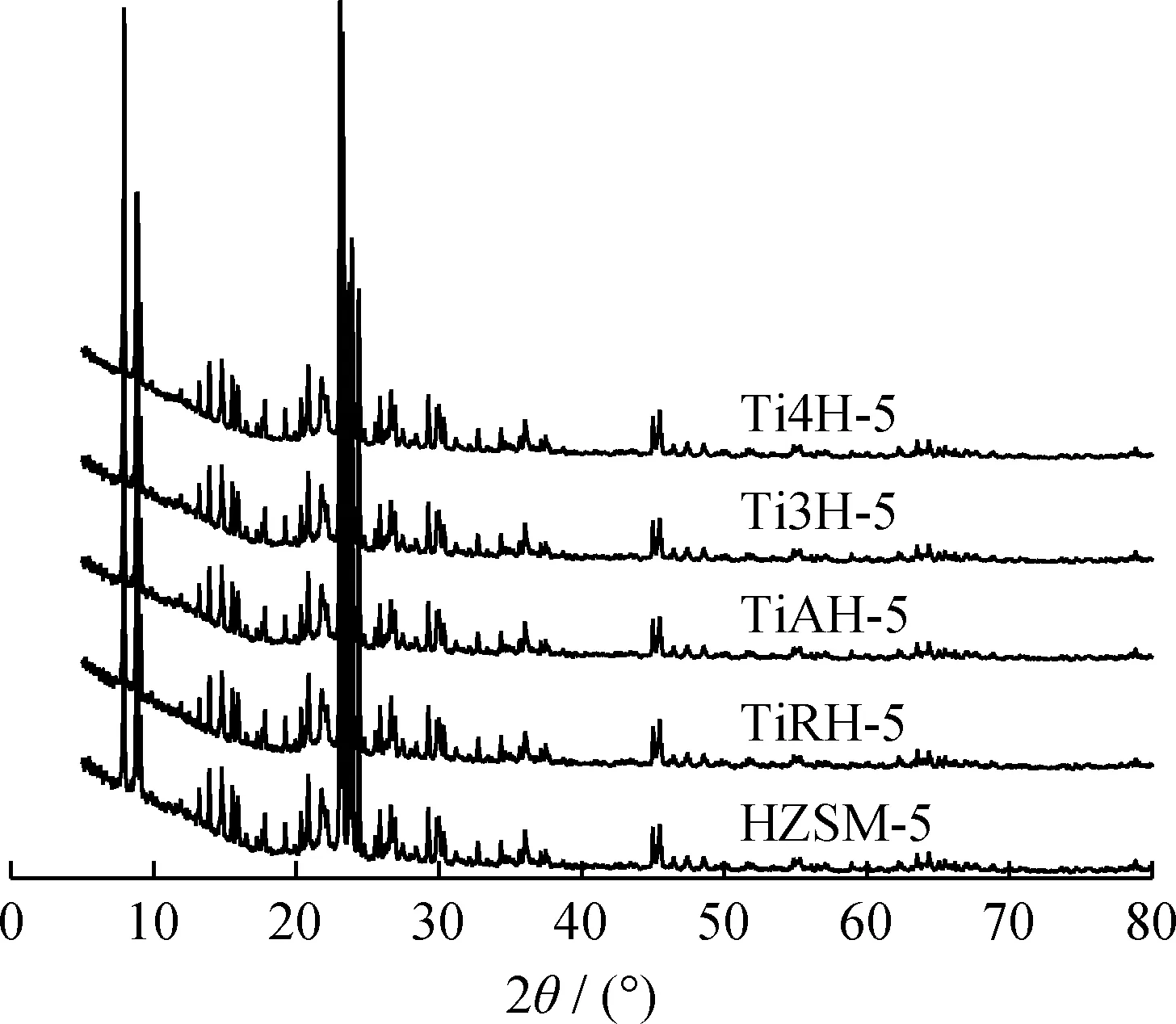
圖2 改性前后催化劑的XRD譜Fig.2 XRD patterns of catalysts before and after modification
2.1.2 催化劑的比表面積和孔體積
鈦改性前后催化劑的比表面積和孔體積參數如表2所示。由表2可見,金紅石型和銳鈦型TiO2改性的TiRH-5和TiAH-5催化劑比表面積及孔體積有所降低;TiCl3和TiCl4改性的Ti3H-5和Ti4H-5催化劑比表面積和微孔孔體積亦降低,但總孔體積明顯增大。這是因為TiCl3和TiCl4改性過程中,溶液中的HCl會去除晶間堆積通道中的非晶態顆粒,使孔徑增大。
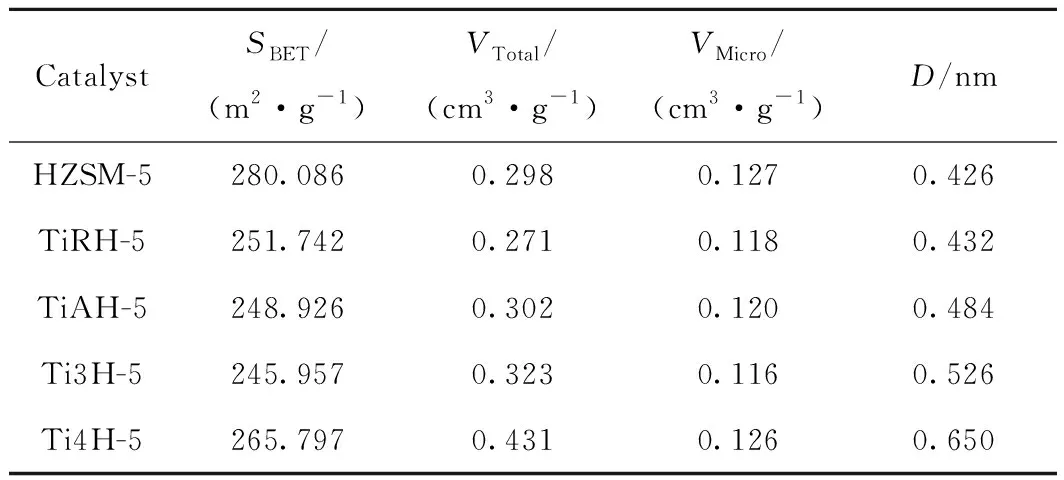
表2 鈦改性前后催化劑比表面積及孔體積Table 2 Specific surface area and pore volume results ofcatalysts before and after modification
2.1.3 Py-IR分析
鈦改性前后催化劑的吡啶紅外脫附譜如圖3所示。其中,波數在1450 cm-1和1616 cm-1附近的吸收峰代表弱L酸中心;1545 cm-1附近的吸收峰代表強B酸中心;而1490 cm-1附近的吸收峰則為弱B酸中心和較強的L酸中心的共同特征峰,這部分酸中心催化作用較弱,一般不進行半定量分析[14]。B酸和L酸酸性的半定量分析結果如表3所示。由表3可見,金紅石型和銳鈦型TiO2改性使催化劑各酸性位點減少,金紅石型TiO2改性降低了催化劑的B/L酸量比,而銳鈦型TiO2改性提高了催化劑的B/L酸量比。在用TiCl3和TiCl4改性過程中,催化劑經過鹽酸處理,使B酸酸量明顯增加;同時氫離子與金屬離子的交換,使部分B酸中心向L酸中心轉化[15];此外,催化劑骨架Al能與金屬離子作用也產生L酸中心[16],使催化劑的L酸中心增加顯著。因此,用TiCl3和TiCl4改性,催化劑的B酸和L酸中心都增加,但n(B)/n(L)降低。
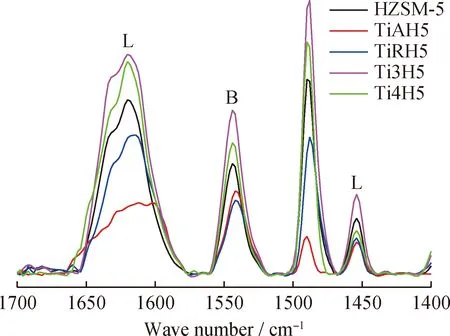
圖3 鈦改性前后催化劑吡啶紅外脫附譜Fig.3 Py-IR desorption spectra of catalystsbefore and after modification
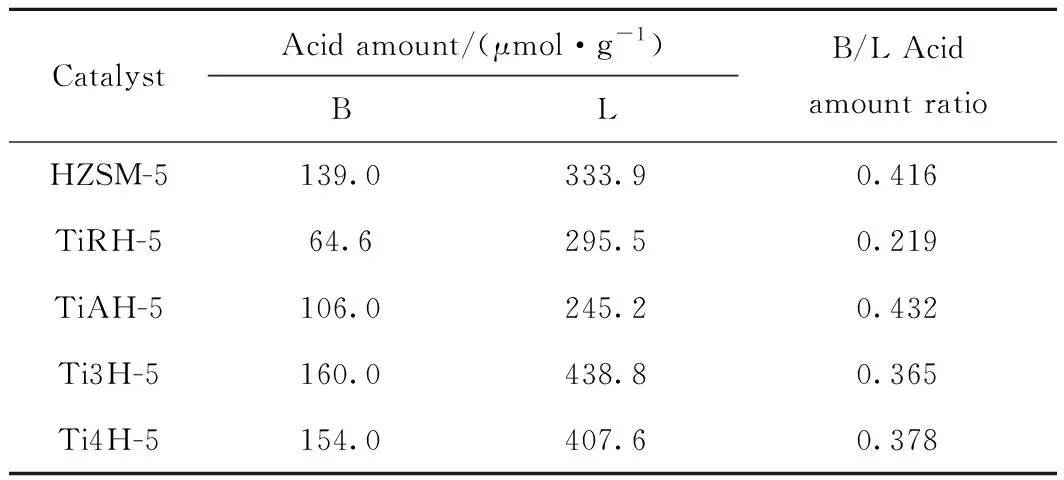
表3 吡啶紅外半定量鈦改性前后催化劑上酸量Table 3 Py-IR semi-quantitative results of catalystsbefore and after modification
2.2 在線光譜分析
使用不同鈦源改性催化劑時反應體系的在線光譜分析結果如圖4所示。由圖4可見:位于431 nm位置的發射線歸屬于CH自由基[17];位于486 nm和656 nm位置的發射線歸屬于Hβ和Hα原子自由基[18];位于516 nm和590 nm位置的發射線分別對應C2烴和C自由基[19];而位于777 nm和844 nm位置的發射線則歸屬于高活性O自由基[18]。一般而言,譜峰強度越大表明自由基濃度越高,因此可以將譜峰強度作為半定量分析依據。當以TiRH-5、TiAH-5和Ti4H-5為催化劑時,主要自由基的發射線強度較用HZSM-5時變化不大,甚至還有所減弱。這是因為銳鈦型、金紅石型以及常規TiO2中鈦元素的價態及鍵構較穩定,與CPD的協同作用較弱,難以產生更多活性自由基;同時,由于改性物掩蓋了HZSM-5部分酸性位點,削弱了催化劑酸性位點與CPD的協同作用。而當以Ti3H-5為催化劑時,大部分自由基的發射線強度較高,原因在于用TiCl3改性引入了中間價態的鈦,其傾向失去電子而被進一步氧化,消耗O自由基,從而促進了CPD誘導的生成O自由基反應,而O自由基增多會促使長鏈大分子有機物斷鏈分解,產生更多的H和C自由基[20]。此外,CPD誘導過程中釋放的高能電子會還原被O自由基氧化的鈦,使中間價態的鈦處于被氧化、還原的動態反應過程中,從而促進自由基反應。因此,以TiCl3改性的催化劑Ti3H-5對反應體系中生成活性自由基的促進作用最強。
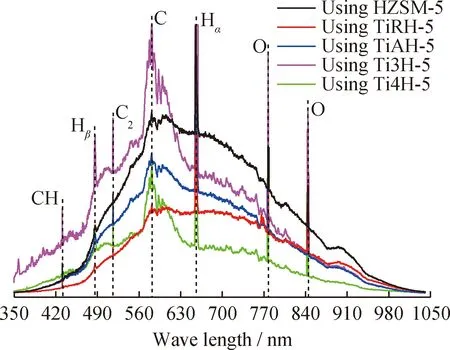
圖4 不同鈦源改性催化劑催化反應體系的在線波譜Fig.4 Online spectra of reaction system with differentTi sources modified catalysts
2.3 精制生物油分析
2.3.1 精制生物油產率及元素組成
采用不同鈦源改性催化劑耦合CPD技術的生物油提質反應中,提質精制生物油的產率、元素組成、有效氫碳比(nH/nC)[12]和HHV見表4。由表4可見,與RB(0)相比,RB(R)產率提高但HHV降低,主要原因在于HZSM-5負載金紅石型TiO2的改性催化劑TiRH-5活性下降,導致生物油提質反應進行不徹底,保留了很多含氧有機物;雖然表觀產率升高,但改性效果很差。
RB(A)的產率最高,為14.57%;其nH/nC和HHV均明顯升高。這是因為銳鈦型TiO2是一種高性能光催化劑,由其改性的TiAH-5催化劑在耦合CPD技術時會產生驅動TiO2光催化作用的紫外光[21]。光生電子易被含氧有機物捕捉,促使有機物的活化或分解,形成更多的碳氫碎片;而空穴則更易吸附和轉化有機物和活性自由基[22]。此外,TiAH-5催化劑L酸中心的脫氫作用較強,銳鈦型TiO2改性使TiAH-5的n(B)/n(L)升高,生物油提質反應傾向于保留氫元素,使高nH/nC的烴含量增加。
RB(3)的產率較高,其nH/nC和HHV提高最明顯。這主要得益于Ti3H-5催化劑上中間價態的鈦處于“氧化-還原”的動平衡狀態,有利于提升自由基反應效率,產生更多的H和C自由基,促進生物油催化提質反應[9]。與RB(0)相比,RB(4)的產率降低,nH/nC和HHV略有升高,但不明顯;RB(4)與RB(A)和RB(3)相比,其產率、nH/nC和HHV均顯著降低,表明常規TiO2與CPD之間亦無明顯的協同作用。因此,以TiCl3改性的催化劑Ti3H-5能同時提升精制生物油產率和燃料品位。
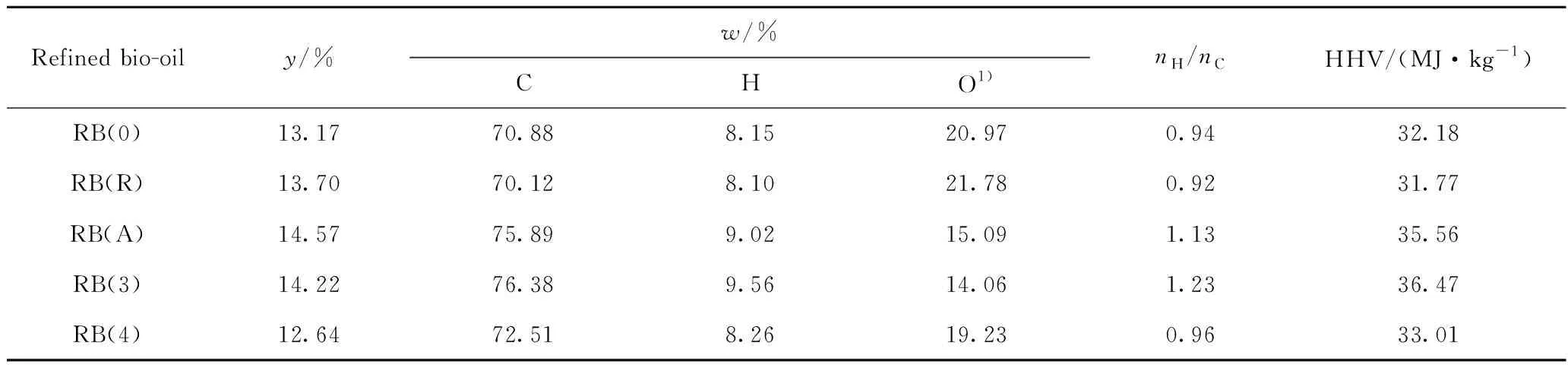
表4 精制生物油產率及元素組成Table 4 Yields and elemental compositions of refined bio-oils
2.3.2 精制生物油的組成
生物油提質精制產物主要為烴類化合物和含氧化合物。其中,烴類化合物主要有單環芳香烴(MAHs)、多環芳香烴(PAHs)和輕質脂肪烴(LAHs);含氧化合物主要有呋喃類(FUR)、酚類(PHE)、醇類(ALC)、醛/酮類(ALD/KET)和酯/醚類(EST/ETH)化合物。
不同催化劑催化生物油提質反應的產物組成如表5所示。由表5可見:與RB(0)相比,RB(R)中MAHs和PAHs含量有所降低,而LAHs有所增加,烴類化合物總含量降低,表明TiRH-5的芳構化性能變差;含氧化合物除了FUR含量降低之外,其他均不同程度增加,而FUR的減少是由于TiRH-5的n(B)/n(L)降低導致部分呋喃開環生成了羥基[23]。
RB(A)的烴類化合物總含量升高,氧含量降低,使HHV升高;而烴類含量升高又以LAHs和MAHs增加為主,其氫含量相對較高,因而使產物的nH/nC升高;同時,RB(A)中含羰基的ALC和ALD/KET含量均較低,主要是因為羰基氧的電負性很強,在銳鈦型TiO2的光催化作用下被捕獲并附著在光生空穴上,空穴將其轉化為水或碳氧化物,進而除去產物中氧[22]。
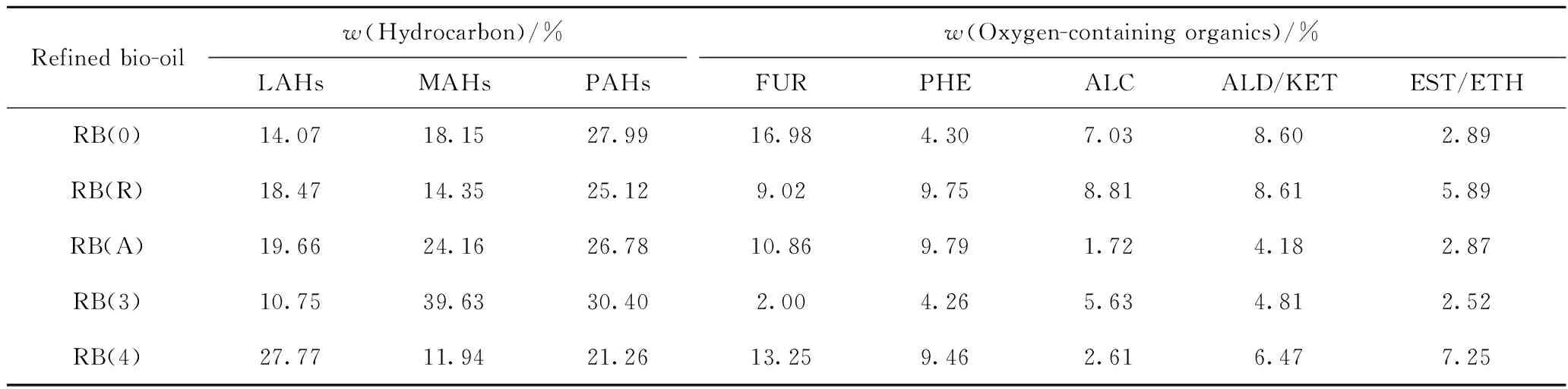
表5 不同催化劑催化精制生物油的產物組成Table 5 Components of refined bio-oils catalyzed with different catalysts
RB(3)中烴類化合物總含量增加,MAHs顯著增加,PAHs稍有增加,而LAHs明顯減少;各類含氧化合物含量均明顯降低。主要原因有2點:一是中間價態鈦元素消耗更多的O自由基,產生更多的H和C自由基參與催化反應,使含氧化合物減少、烴類增加;二是B/L酸量比降低,即L酸中心相對數量升高,促進環烷烴脫氫生成芳環。
RB(4)的烴類化合物總含量略有增加,并以LAHs增加為主,原因在于改性后的催化劑上具有較強的裂解脫氫作用的L酸中心顯著增加、但具有成環作用的B酸中心減少;含氧有機物中FUR、ALC和ALD/KET含量有不同程度降低,PHE和EST/ETH含量則明顯升高,表明Ti4H-5的脫氧催化能力有限,部分含氧有機物轉化并未形成烴類,而是轉化了其他含氧有機物。
因此,以TiCl3改性的催化劑Ti3H-5對烴類化合物的選擇性最強,對含氧有機物的轉化能力最好。
對精制生物油中烴的碳數分布進行分析,結果如表6所示。以汽油(C4~C12)和柴油(C10~C22)的碳數分布為分界依據,將精制生物油中烴化合物的碳數分布劃分為C4~C、C10~C12和C13~C22等3個區域。精制生物油中烴碳數均在C7~C19范圍內,符合燃料的碳數要求。由表6可知,與RB(0)相比,RB(R)碳數分布變化不大;RB(A)中C4~C9烴明顯增加,C4~C12烴的質量分數為47.34%,可作為汽油添加劑或替代組分;而RB(3)中C10以上的烴顯著增加,C10~C22烴質量分數達76.42%,得益于芳香烴含量的大幅增加,可作為柴油添加劑或替代組分;RB(4)中3個區域的烴質量分數相對均衡,說明Ti4H-5對芳香烴的選擇性降低,而對LAHs烴選擇性升高。
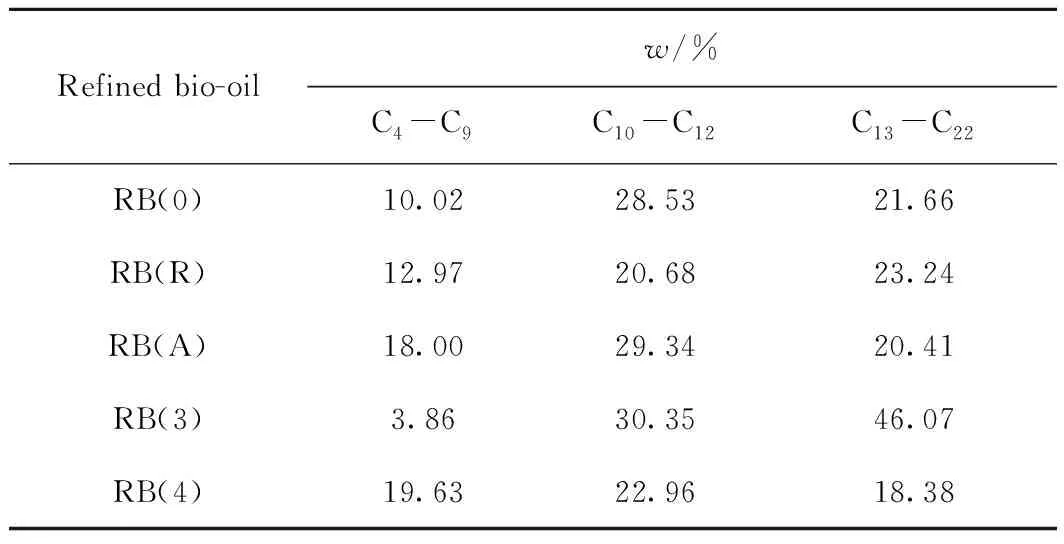
表6 烴類化合物的碳數分布Table 6 Carbon atom distribution of hydrocarbonsin bio-oil products
因此,銳鈦型TiO2改性的催化劑TiAH-5催化生物油提質,可以得到較高產率的汽油添加劑或替代組分;以TiCl3改性的催化劑Ti3H-5催化生物油提質,可以得到較高產率的柴油添加劑或替代組分。
2.4 改性催化劑抗結焦性能分析
2.4.1 TG-DTG分析
反應后催化劑的TG和DTG曲線如圖5所示。由圖5(a) TG曲線可知:使用后催化劑在低于 250 ℃ 的質量損失主要為催化劑上吸附的水分和輕質小分子化合物;250~700 ℃內的質量損失由催化劑上的焦炭氧化分解引起。對DTG曲線進行Gaussian分峰擬合,計算得到HZSM-5、TiRH-5、TiAH-5、Ti3H-5和Ti4H-5的總結焦量依次為7.93%、4.56%、2.81%、1.76%和6.29%,因此,催化劑穩定性順序由高到低依次為Ti3H-5、TiAH-5、TiRH-5、Ti4H-5、HZSM-5。
一般而言,生物油催化提質后催化劑上會產生2種類型的焦炭:低溫下結焦的含氧型焦炭和高溫下結焦的純碳型焦炭。含氧型焦炭多為絲狀,多分布于催化劑表面,而純碳型焦炭無固定形狀,多位于催化劑孔道內[24]。對于Ti3H-5,由于中間價態的鈦與CPD之間的相互作用,使含氧型焦炭難于在其上結焦,因此反應后Ti3H-5上的焦炭為純碳型焦炭,分解溫度相對較高。對于TiAH-5,由于銳鈦型TiO2的光催化作用有利于羰基的轉化和去除,減少了含氧型焦炭的生成;同時,銳鈦型TiO2改性使催化劑的B/L酸量比升高,傾向于保留氫元素,使高氫/碳比烴類增加,減少了純碳型焦炭,因此反應后TiAH-5上的焦炭較少。含氧型焦炭分解溫度相對偏低,而純碳型焦炭分解溫度偏高[12]。對于HZSM-5、TiRH-5和Ti4H-5,反應后焦炭主要由低溫含氧型焦炭和少量高溫純碳型焦炭組成[25]。
2.4.2 TEM分析
反應后催化劑的透射電鏡照片如圖6所示。由圖6可見,反應后HZSM-5結焦量較高而使催化劑透明度較低,其表面明顯附著絲狀含氧型焦炭。反應后TiRH-5和Ti4H-5的透明度與HZSM-5接近,說明催化劑表面焦炭含量較高。TiRH-5上未觀察到明顯的絲狀含氧型焦炭,而Ti4H-5表面觀察到明顯絲狀含氧型焦炭,說明常規TiO2與CPD之間缺乏相互作用。銳鈦型TiO2的光催化作用使TiAH-5結焦量下降,使TiAH-5顆粒上部分區域的透明度提高。對于反應后Ti3H-5,只有少量焦炭
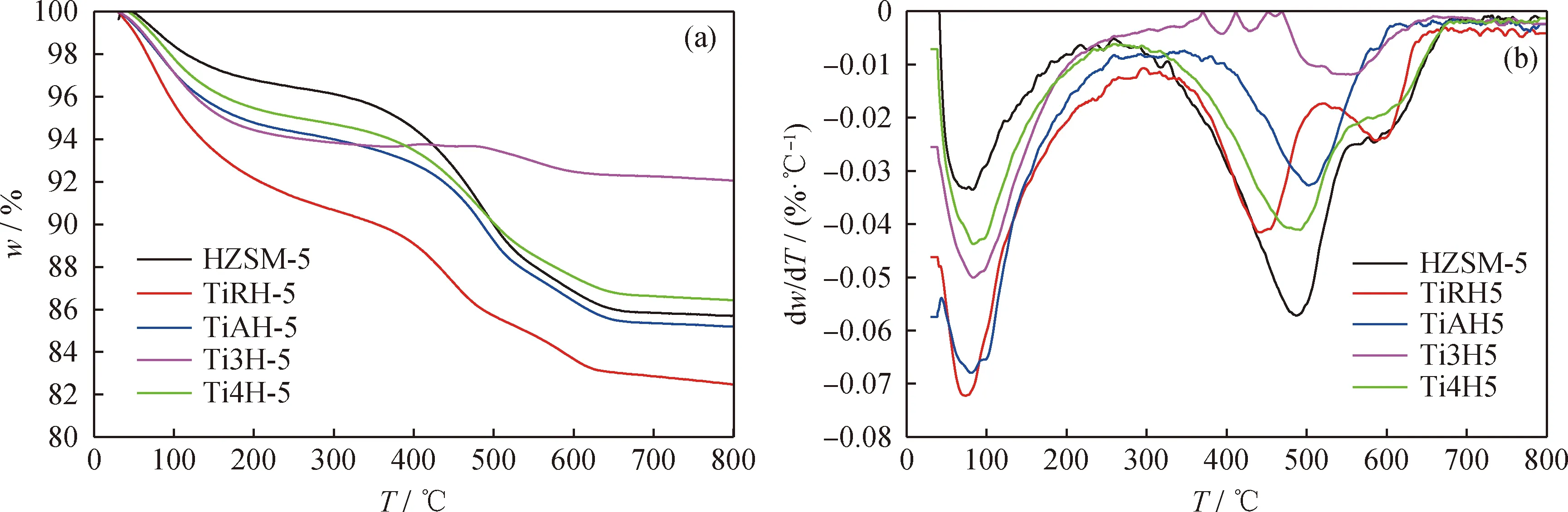
圖5 反應后催化劑的TG和DTG曲線Fig.5 TG and DTG profiles of catalysts after reaction(a) TG; (b) DTG

圖6 反應后催化劑的透射電鏡照片Fig.6 TEM images of different catalysts after reaction(a) HZSM-5; (b) TiRH-5; (c) TiAH-5; (d) Ti3H-5; (e) Ti4H-5
點綴于催化劑顆粒上。這是因為中間價態鈦元素與CPD的相互作用,使鈦處于“氧化-還原”的動平衡狀態,提升自由基反應效率,使O自由基增多,有利于分解焦炭前驅物,因此Ti3H-5上結焦較少。
3 結 論
以銳鈦型TiO2改性的TiAH-5耦合CPD可以提升精制生物油產率至最高值14.57%,但對高位熱值的提升作用不是最顯著,精制生物油中 C4~C12烴質量分數為47.34%,較適合制成汽油添加劑或替代組分。
以TiCl3改性的Ti3H-5耦合CPD對生物油的催化提質性能最優,精制生物油產率和HHV分別達到14.22%和36.47 MJ/kg,烴質量分數達到了80.78%,其中C10~C22烴質量分數達76.42%,適合用作柴油添加劑或替代組分。TiCl3改性引入的中間價態鈦元素與CPD相互作用使Ti3H-5結焦量最低,穩定性最好。
以金紅石型TiO2改性催化劑TiRH-5性能變差。負載常規TiO2改性催化劑Ti4H-5酸性增強,但與CPD無協同作用,催化劑性能變化不明顯。
猜你喜歡
天天愛科學(2022年9期)2022-09-15 01:12:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
航空世界(2020年10期)2020-01-19 14:36:20
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年11期)2015-10-14 01:14:14
