混合型肝豆狀核變性1例報告
2021-03-08 09:08:20張碩,朱輝,呂洋
中風與神經疾病雜志 2021年2期
關鍵詞:癥狀
張 碩, 朱 輝, 呂 洋
肝豆狀核變性(hepatolenticular degeneration,HLD)又稱為Wilson病(Wilson’s disease,WD),是一種常染色體單基因隱性遺傳的銅代謝障礙性疾病,由于編碼銅跨膜轉運蛋白的基因ATP7B發生突變,以致銅在肝臟、大腦及其他組織器官中過度沉積。多數HLD患者以肝病和神經精神癥狀起病,常累及多個器官,最初的癥狀往往不易與其他神經系統疾病相鑒別,易誤診[1]。現報道我科收治的1例臨床上較少見的以運動功能障礙為首發癥狀的混合型肝豆狀核變性患者,以提高對該病的認識。
1 臨床資料
患者,男,28歲,因“言語不清伴行走不穩13 y,加重伴肢體扭轉9 m”于2019年12月入院。患者于入院前13 y家人發現其講話不如以往清晰、流利,同時出現行走緩慢、不穩,先后就診于多家醫院未明確診治,后就診于我院行頭部CT,眼科檢查雙眼K-F環(+),血清銅藍蛋白降低且24 h尿銅含量升高,診斷為“肝豆狀核變性”,給予驅銅、護肝、營養神經等治療,期間因脾大、脾亢行“脾切除術”治療。2008年初,因未遵醫囑服用藥物,逐漸出現脾氣暴躁、易怒,動輒打人毀物,加用“氯丙嗪”控制精神癥狀。2011年后相繼自行停用氯丙嗪、驅銅等藥物,病情尚穩定。9 m前,患者言語不清伴行走不穩癥狀再次加重,且出現肢體扭轉伴疼痛。發病以來,飲食、睡眠尚可,二便如常。父母體鍵,否認近親結婚史,患者一哥哥產后全身皮膚黝黑,出生3 d死亡(未查病因)。家族成員中否認類似病史。內科查體:全身皮膚黏膜及雙鞏膜無黃染,腹平軟,腹中部可見一長約20 cm陳舊性手術疤痕,肝肋下未觸及,脾已切除,移動性濁音(-),雙下肢無水腫。神經系統查體:神志清楚,說話緩慢而含糊,發音單調,發音費力,僅可簡單應答。雙眼球活動自如,無眼震。伸舌不能,雙側軟腭抬舉減弱,軟腭反射遲鈍。左上肢、雙下肢屈曲扭轉畸形。四肢肌力5級。四肢肌張力呈鉛管樣增高。雙側病理反射未引出。余查體不合作。
輔助檢查:肝功:門冬氨酸氨基轉移酶50.9 U/L;腎功:肌酐114.5 μmol/L;血脂:甘油三脂1.81 mmol/L,高密度脂蛋白0.94 mmol/L,低密度脂蛋白3.75 mmol/L;銅生化:銅藍蛋白33.7 mg/L,銅1.3 μmol/L,銅氧化酶0.021OD。24 h尿銅175.1 μg。頭部CT(見圖1):雙側蒼白球區、橋腦低密度影。腹部彩超:(1)肝豆肝病樣改變(結節型);(2)膽囊炎;(3)右腎結石。腹腔血管彩超:門靜脈主干內未見明顯血栓形成。眼部檢查:K-F環(+)。基因檢測:ATP7B基因p.R778L雜合突變,一個意義未明的內含子區剪切突變(見圖2)。肌電圖:可見左肱二頭肌、尺側腕屈肌、指短伸肌及右拇長伸肌成組中量MUP陣發性發放。UWDRS評分量表:神經功能評分112分,精神癥狀評分22分。診斷:肝豆狀核變性(混合型),ATP7B基因p.R778L雜合突變。患者在外院進行驅銅治療,1 m后癥狀好轉,但未能按療程完成治療,1 y后電話隨訪,患者癥狀再次加重,建議其系統治療。
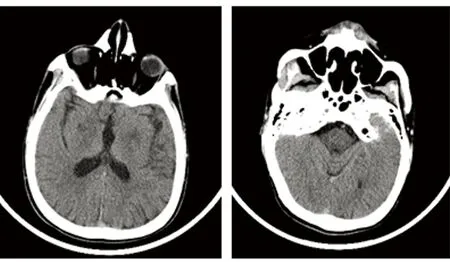
圖1 雙側蒼白球區、橋腦低密度影

圖2 ATP7B基因p.R778L雜合突變
2 討 論
英國神經病學家Samuel Wilson[2]于1912年首次發現肝豆狀核變性,是位于13號染色體的ATP7B基因突變,使其編碼的P型ATP酶功能減弱甚至消失,致膽道排銅障礙及血清銅藍蛋白合成減少,大量銅沉積在不同的組織中,典型癥狀為進行性加重的神經系統表現、肝臟疾病和眼部改變。
2008年《肝豆狀核變性診斷與治療指南》[3]將肝豆狀核變性分為4型:腦型、肝型、其他類型及混合型。Wilson病主要累及中樞神經系統稱為腦型Wilson病,多數表現為運動功能障礙,比如原因不明的錐體外系癥狀,特別是手部遠端不對稱性震顫或頭部、軀干震顫、肌張力增高、共濟失調等,也可出現流涎、構音障礙、吞咽困難,嚴重者會出現性格改變、精神分裂等精神癥狀。其中將腦型WD又劃分為典型WD型、扭轉痙攣型、精神障礙型及假性硬化型。WD發病年齡通常>15歲,也有報道為7~9歲[4],本例患者發病時16歲,以構音障礙、共濟失調為首發癥狀,逐漸出現精神癥狀,精神癥狀是非特異性的,可以表現為抑郁、躁狂,或僅僅為情緒不穩定。故當青少年患者出現不能解釋的認知障礙、精神障礙或者運動障礙需排除此病。
肝臟發生病理改變為主的稱為肝型Wilson病,表現為肝炎、肝硬化及肝衰竭等臨床癥狀。K-F環[5],即Wilson病患者角膜處黃棕色或黃綠色色素環,是由于銅顆粒沉積于角膜后彈力層形成的,幾乎見于所有出現神經病變的患者中,為該病最常見的眼部體征,但K-F環陰性也不能排除WD的可能。此外,也可造成腎臟損傷、骨關節肌肉損害或溶血性貧血,屬于其他類型Wilson病。混合型WD是以上各型的組合。
本例患者為青年男性,具有肝豆狀核變性經典的“三聯征”,累及中樞神經系統、肝臟及眼部,且病情持續進展。特別之處在于,本例患者以神經損傷為突出表現(腦型),同時肝實質改變(肝型),肌酐升高提示腎臟損害(其他類型),為臨床比較少見的混合型Wilson病。目前國內外有關混合型WD報道較少,呂佳等[6]2019年報道了以神經障礙為突出癥狀的肝豆狀核變性1例,此患者以神經損傷為突出表現,同時存在肝硬化,尿常規見隱血、肌酸激酶升高提示腎臟、骨骼肌損害。神經系統表現具有較大的臨床異質性,最常見的神經系統異常包括構音障礙、肌張力障礙、震顫和帕金森病,疾病的非典型表現增加了診斷的復雜性。患者肝實質發生了肝豆肝病樣改變(結節型),肝功化驗中門冬氨酸氨基轉移酶略有異常,考慮與患者長期驅銅、服用護肝片等對癥治療有關;患者肌酐高于正常,說明銅對腎臟也造成了一定程度的損傷。銅藍蛋白是一種主要由肝臟表達合成的含銅的α2球蛋白,運輸血液中90% 以上的銅至各個器官,參與人體內銅、鐵等微量元素的平衡,患者的銅藍蛋白及血清銅、尿酮均發生相應改變。
對該患者進行了基因檢測,第8號外顯子測序顯示,ATP7B基因編碼區檢出c.2333G>T雜合突變,是我國肝豆狀核變性患者常見基因突變位點。HLD患者的基因水平確診需滿足ATP7B基因純合致病突變或復合雜合致病突變,單一雜合突變不能確診[7]。本患者ATP7B基因不符合確診條件,其患者父母也未能行基因檢測,但全球發現WD患者ATP7B基因突變類型700余種,少數屬于內含子突變,尤其是剪切突變,故只檢測突變熱點用于診斷和篩查,容易漏診[8],而且WD診斷是從基因到臨床的綜合統一,不是靠單一指標。本患者基因檢測出4號內含子區c.1708-1G>C剪切突變,但意義未明,提示我們需要對更多的家系進行基因研究,以便更全面地了解WD患病情況。
本研究報道的病例應該引起對HLD的不同分型及起病形式多樣更充分的認識,臨床醫生應仔細詢問病史,同時檢查血清銅藍蛋白、血銅、24 h尿銅、角膜K-F環及ATP7B基因等多項指標,減少誤診、漏診,Wilson病屬于目前少數幾種可控的神經遺傳病之一,使患者得到早診斷、早治療,最終改善疾病預后。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
