線粒體質量控制失調與特發性肺纖維化的關系研究進展
2022-04-01 08:10:16馮同高瑕王波李萬成
解放軍醫學雜志 2022年1期
馮同,高瑕,王波,李萬成*
1成都醫學院,成都 610500;2成都醫學院第一附屬醫院呼吸與危重癥醫學科,成都 610500
特發性肺纖維化(idiopathic pulmonary fibrosis,IPF)是一種嚴重影響肺通氣與換氣功能的慢性下呼吸道疾病,表現為異常的肺間質纖維化及炎癥,包括肺泡結構的破壞。IPF多發生于中年及老年人,平均診斷年齡為66歲,中位生存期為確診后3~5年。IPF年發病率為(4.6~16.3)/10萬,且呈上升趨勢;患病率為(13~20)/10萬,男性患者比例較高(男:女=1.5~1.7:1)。關于IPF的發病機制,目前被較多學者支持的是上皮-間充質轉化(epithelial-tomesenchymal transition,EMT)。IPF被認為是肺泡上皮細胞功能異常及凋亡的結果,該損傷引起間充質細胞的遷移、增殖、活化,并形成了成纖維細胞及肌成纖維細胞病灶。激活的肌成纖維細胞分泌過多的細胞外基質,破壞了肺部結構[1]。目前,美國食品藥品管理局(Food and Drug Administration,FDA)批準用于治療IPF的藥物有吡非尼酮及尼達尼布,但這兩種藥物的療效有限,僅能適度緩解疾病的癥狀,不能改善IPF患者的生活質量及降低死亡率[2]。
線粒體除了具有能量供給的功能外,還在信號傳導、新陳代謝及細胞死亡的調控中起重要作用[3]。線粒體質量控制通常通過三種不同的機制來維持:(1)線粒體生物合成;(2)線粒體動力學(融合與分裂);(3)線粒體自噬。廣義的線粒體質量控制還包括線粒體內活性氧水平的調控,線粒體基質的蛋白酶、泛素蛋白酶體系統及線粒體衍生的囊泡。機體通過這些機制生成新的線粒體,限制損傷與衰老的線粒體過度積累,減少活性氧(reactive oxygen species,ROS)的產生,從而維持線粒體的動態平衡。本文探討了包括線粒體ROS調控在內的線粒體質量控制失調與IPF的相關性,并針對線粒體質量控制提出IPF的防治策略。
1 線粒體質量控制
線粒體通過線粒體質量控制,即通過融合/分裂改變其形狀及大小,通過線粒體自噬包裹受損的線粒體并將其傳遞至溶酶體進行清除,通過生物合成新生線粒體以補充線粒體池,通過線粒體內抗氧化系統降低ROS對細胞造成的氧化損傷,這些過程相互協調作用以適應不同條件下細胞新陳代謝的變化。
1.1 ROS水平的線粒體質量控制 ROS是一類含活性氧原子的反應性化學物質,包括超氧陰離子、羥基自由基、過氧化氫、臭氧及單線態氧等[4]。過多的ROS累積可造成線粒體的氧化破壞,同時,ROS作為信號分子參與了線粒體質量控制的調節,激活抗氧化系統。細胞內含有的抗氧化系統負責清除過多的ROS,維持氧化還原狀態的穩定,阻止ROS誘導的氧化應激對線粒體DNA(mtDNA)及蛋白質的破壞。
1.2 線粒體動力學對線粒體質量控制的調控
線粒體分裂通過胞質動力相關蛋白1(dynamin relatedprotein 1,DRP1)、分裂蛋白1(fission protein 1,FIS1)、線粒體分裂因子(mitochondria fission factor,MFF)及線粒體動力蛋白49/51(mitochondrial dynamics proteins of 49 and 51,MID49/51)的作用完成。當位于細胞質或內質網中的DRP1被募集到線粒體外膜上時,DRP1與線粒體外膜上的受體FIS、MFF、MID49/51相互作用形成復合物,與DRP1結合的GTP的水解會收縮DRP1,使線粒體膜分裂,導致兩個獨立的子代線粒體形成,從而發生裂變[5]。線粒體外膜融合是通過線粒體融合蛋白(mitofusin 1/2,MFN1/2)及視神經萎縮相關蛋白1(optic atrophy 1,OPA1)進行的。OPA1在YME1樣ATP酶(YME1-like 1 ATPase,YME1L1)的調控下,形成長式及短式的寡聚體,它們之間保持平衡,維持線粒體內膜的融合[6-7]。
1.3 線粒體自噬對線粒體質量控制的調控 線粒體自噬相關蛋白可分為E3泛素連接酶PARKIN依賴型及非PARKIN依賴型。其中最經典的途徑為E3泛素連接酶PARKIN依賴型,需要磷酸酶及張力蛋白同源物誘導的蛋白激酶1(PTEN induced putative kinase 1,PINK1)的參與。正常情況下,PINK1位于線粒體內膜,由線粒體特異性蛋白酶線粒體內膜早老素相關菱形樣蛋白(presenilin associated rhomboid like protein,PARL)及加工肽酶(mitochondrial processing peptidase,MPP)降解[8-9]。在線粒體損傷的情況下,PARL及MPP失活,抑制了PINK1由線粒體外膜轉運至線粒體內膜的進程,導致PINK1在線粒體外膜上累積。位于線粒體外膜的PINK1的激酶結構域不僅可磷酸化線粒體外膜蛋白,還可募集及激活E3-泛素連接酶PARKIN[10]。激活的PARKIN將泛素化線粒體外膜及細胞質蛋白,泛素化蛋白與銜接子蛋白SQSTM1/p62及微管相關蛋白1輕鏈3(MAP1LC3/LC3)結合形成自噬體,啟動自噬機制降解損傷嚴重的線粒體。
1.4 線粒體生物合成對線粒體質量控制的調控線粒體生物合成是一個高度協調的過程,利用線粒體及核編碼基因來改變線粒體的大小及質量。它需要線粒體、細胞核、內質網及其他細胞器的協同作用。線粒體生物合成中關鍵的調控分子是過氧化物酶體增殖物激活受體-γ共激活因子-1α(PGC-1α),但其他轉錄因子,包括5'腺苷單磷酸激活蛋白激酶(5'-AMP activated protein kinase,AMPK)及核呼吸因子1/2(nuclear respiratory factor 1/2,NRF1/2)也參與其中[11]。PGC-1α作為共轉錄因子,轉位至細胞核與NRF1/2結合,轉錄激活線粒體轉錄因子A(mitochondrial transcription factor A,TFAM)的表達,從而驅動mtDNA的轉錄及復制。
2 線粒體質量控制失調與IPF發病的相關性
目前,已有研究證據表明IPF與線粒體質量控制失調密切相關。人們已經認識到線粒體功能障礙是IPF及其他肺部疾病如慢性阻塞性肺疾病、哮喘發病機制的關鍵環節[12-14]。
2.1 線粒體氧化應激在IPF發病中的作用 ROS是參與細胞正常分化、增殖等過程的反應性物質,在病理狀態下,線粒體內產生過多的ROS,不能被抗氧化系統及時清除,就會引發一系列反應,如mtDNA損傷、通透性轉換孔開放及膜通透性改變,導致肺纖維化的發生[15]。研究發現,過量的線粒體ROS(mtROS)產生及蛋白酶復合體Ⅰ、蛋白酶復合體Ⅳ活性下降與纖維化小鼠模型中Ⅱ型肺泡上皮細胞(alveolar epithelial cells,AEC)的線粒體功能障礙有關[16-17]。與野生型小鼠相比,線粒體過氧化氫酶高表達轉基因小鼠接觸石棉、博萊霉素后肺纖維化的程度降低,小鼠Ⅱ型AEC中的mtROS水平也降低[17]。NRF2是一種氧化劑敏感的轉錄因子,可通過激活抗氧化系統,調節谷胱甘肽、硫氧還蛋白和還原型煙酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)的生物合成、利用及再生,并能控制線粒體及NADPH氧化酶的ROS生成,在維持細胞氧化還原穩態中起著至關重要的作用[18]。NRF2基因敲除小鼠肺部多種抗氧化酶表達降低,從而易發生ROS誘導的肺纖維化損傷[19]。同時,mtDNA損傷與氧化損傷及肺纖維化有關。IPF患者中mtDNA活性氧水平上升[20]。DNA堿基切除修復酶8-氧鳥嘌呤DNA糖基化酶1(8-oxoguanine DNA glycosylase 1,Ogg1)對修復Ⅱ型AEC氧化損傷起重要作用[21]。與野生型小鼠相比,Ogg1基因敲除小鼠石棉暴露后的肺纖維化程度、Ⅱ型AEC mtDNA損傷及氧化劑誘導的細胞凋亡增加[22]。線粒體哺乳動物的沉默信息調節因子3(silent information regulator 3,SIRT3)是煙酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD)依賴的sirtuin家族的成員,參與肺纖維化的調節。Sirt3基因敲除衰老小鼠會發生多個器官的組織纖維化,包括心臟、肝臟、腎臟及肺部,且IPF患者肺內SIRT3的活性下降[23]。SIRT3通過Ogg1及錳超氧化物歧化酶(Mn superoxide dismutase,MnSOD)調節蛋白脫乙酰化來減少mtDNA損傷,SIRT3缺乏可導致線粒體蛋白的乙酰化及失活,增強ROS引起的mtDNA損傷及細胞凋亡,促進肺纖維化[23]。因此,線粒體ROS增多是肺纖維化病理改變的機制之一。
2.2 線粒體動力學在IPF發病中的作用 線粒體是動態的細胞器,通過不斷的融合與分裂,交換損傷的蛋白質及mtDNA,保持著動態平衡。小鼠Ⅱ型AEC中缺乏MFN1及MFN2會導致自發性肺纖維化的發生率及病死率增高。MFN1缺失導致線粒體破碎,MFN2缺失導致線粒體腫脹。在暴露于博來霉素的小鼠中,Ⅱ型AEC細胞的MFN1、MFN2及DRP1的表達水平增高,以減輕肺纖維化損傷[24]。G蛋白偶聯受體C族5型A組(G protein-coupled receptor C group 5 type group A,GPRC5A)是G蛋白偶聯受體家族的一個成員。在IPF損傷AEC中,GPRC5A的減少會導致線粒體融合蛋白MFN2及OPA1缺失,從而增加博來霉素所致肺纖維化的膠原蛋白沉積,刺激成纖維細胞的增殖與激活[25]。線粒體的融合及分裂參與了細胞凋亡的調節,線粒體融合可抑制細胞凋亡,而線粒體分裂可促進細胞凋亡。Ⅱ型AEC的凋亡是肺纖維化發生發展的關鍵因素。研究表明,miR-30a表達上調可減少Ⅱ型AEC的凋亡,抑制線粒體分裂,并降低DRP1的表達及轉運,減輕肺纖維化的程度[26]。上述研究提示,線粒體動力學與肺纖維化之間存在密切關系,線粒體動力學改變可能通過影響線粒體活性氧生成、mtDNA分布、細胞內鈣離子穩態及細胞凋亡等影響細胞能量供給及信號轉導,從而促進肺纖維化的發生發展。
2.3 線粒體自噬在IPF發病中的作用 線粒體自噬是降解大分子蛋白質或細胞器的過程。當線粒體受損時,自噬體可將其包裹分隔,并遞送至溶酶體進行清除及再回收[27]。PINK1缺乏會導致線粒體腫脹、功能障礙,并促進衰老相關的肺纖維化[12]。博萊霉素誘導的PARKIN基因敲除小鼠的線粒體自噬不足,無法清除受損線粒體,肺纖維化程度增加[28]。在IPF成纖維細胞灶的肌成纖維細胞和肺成纖維細胞中,PARKIN表達水平降低[29]。轉錄激活因子3(activating transcription factor 3,ATF3)是PINK1基因表達的負調控因子,其過表達可抑制PINK1啟動子的活性,導致去極化線粒體的積累,線粒體ROS的產生增加。在ATF3缺失小鼠中,博來霉素誘導的肺纖維化程度減輕[30]。總之,目前動物實驗及人體研究結果都表明促進自噬可以預防肺纖維化。
2.4 線粒體生物合成在IPF發病中的作用 線粒體生物合成是一個調控新生線粒體形成的生物學過程,主要涉及mtDNA的復制、mtDNA編碼蛋白的合成以及協調線粒體動力學等作用。
線粒體生物合成能力下降與IPF的發病機制有關。在IPF患者與博萊霉素誘導的肺纖維化模型中,PGC-1α表達水平降低,此外,PGC-1α基因敲除小鼠經博萊霉素誘導后肺纖維化程度加重[31]。甲狀腺激素(thyroid hormone,TH)可通過改善線粒體功能及形態、恢復線粒體膜電位來減輕博來霉素誘導的肺纖維化。IPF患者肺組織中碘甲腺原氨酸脫碘酶Ⅱ(recombinant deiodinase iodothyronine typeⅡ,DIO2,一種激活TH的酶)的活性及表達水平高于對照組,且與疾病嚴重程度相關。DIO2基因敲除小鼠經博來霉素誘導后肺纖維化程度加重。在博來霉素暴露后,TH促進了體內及體外AEC中的線粒體生物合成,改善了線粒體生物能,并減少了線粒體調節的細胞凋亡。在PGC-1α或PINK1基因敲除小鼠中,TH不能抑制肺纖維化,提示TH的作用是依賴于PGC-1α和PINK1而實現的[32]。AMPK是細胞生物能的關鍵傳感器,可控制生物體內合成代謝的轉換。在IPF患者及IPF小鼠模型中,AMPK活性在代謝活躍且凋亡抵抗的成纖維細胞相關的纖維化區域較低。AMPK激活物AICAR可上調TFAM并增加線粒體電子傳遞鏈復合物主要成分NADH泛醌脫氫酶1β亞復合物8的表達,增強線粒體的生物合成[33]。目前認為線粒體生物合成受損導致線粒體質量降低是肺纖維化線粒體功能障礙的另一重要內在機制。
3 以線粒體質量控制為靶點的IPF干預策略
恢復線粒體質量控制的干預措施可能是IPF的保護策略,如MitoQ作為線粒體靶向抗氧化劑,可減弱IPF肺成纖維細胞中TGF-β1及NOX4的表達[34]。研究表明,17β-雌二醇通過細胞核或線粒體雌激素受體對線粒體功能的調節可誘導抗氧化反應以及NRF1/2、PGC-1α的激活,從而促進線粒體的生物合成[35]。眾所周知,IPF的AEC中PINK1缺乏是引發線粒體功能障礙的主要因素。因此,增強PINK1活性的藥物可作為IPF的一種治療選擇。三磷酸激動素(kinetin triphosphate,KTP)可增強PINK1、PARKIN的活性并降低細胞凋亡標志物的表達,是抗肺纖維化的潛在藥物[36]。同樣,線粒體蛋白SIRT3對纖維化也有保護作用。Hexafluoro可在博來霉素暴露的小鼠中誘導SIRT3的表達,通過抑制TGF-β1降低膠原蛋白、纖連蛋白及α-平滑肌肌動蛋白(α-SMA)的表達,從而減輕肺纖維化[37]。此外,線粒體質量控制還依賴于酶泛素化的存在來清除受損的線粒體蛋白,該過程受到去泛素化酶(deubiquitinase,DUBs)的負調控,后者通過去除PARKIN添加的泛素標簽來抑制線粒體自噬。USP30(ubiquitin-specific protease 30)是去泛素化酶家族中的一員,去泛素化USP30可以增強線粒體自噬,代表一種新的治療靶點[38-39]。綜上所述,針對線粒體質量控制的藥理學調控以生物合成、線粒體自噬及分裂/融合為目標,從而恢復線粒體功能,最終預防肺纖維化的發生(表1)。
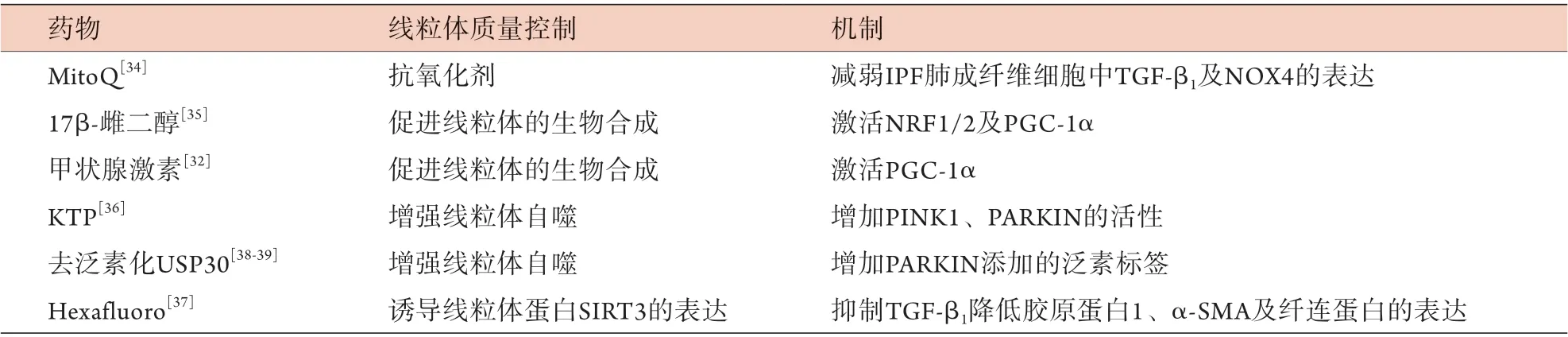
表1 線粒體質量控制干預IPF的現有潛在靶點Fig.1 Current potential targets for mitochondrial quality control in IPF intervention
4 總結與展望
在IPF背景下,線粒體質量控制失調導致線粒體功能障礙,ROS產生增加,誘發細胞凋亡。線粒體質量控制系統(線粒體自噬、線粒體生物合成及線粒體動力學)在IPF中起著不同的作用。IPF中線粒體自噬減少,受損的線粒體降解減少,功能障礙的線粒體聚積。IPF中線粒體的生物合成減少,無法補充新的線粒體成分,因而難以維持及恢復線粒體的數量與功能。TH對PGC-1α的上調作用逆轉了博來霉素誘導的肺纖維化,這歸因于線粒體生物合成增加,從而恢復了線粒體的功能。因此,在IPF中促進線粒體自噬及生物合成可能具有抗纖維化作用。線粒體融合及分裂不是兩個獨立的過程,而是兩個相互依賴的過程。不同的壓力應激環境能夠決定線粒體動力學響應的方式。在輕度到中度應激環境下,線粒體融合將受損的線粒體與正常的線粒體結合起來以防止損傷,將多個線粒體融合為一個線粒體,使線粒體免于自噬。在嚴重的應激環境下,線粒體分裂將受損的線粒體區域與正常的線粒體分開,從而防止了進一步的損傷,受損的線粒體最終通過線粒體自噬去除。IPF中增強的線粒體動力學通過不斷的融合與分裂,交換損傷的蛋白質及mtDNA,保持線粒體的動態平衡。
對于進行性發展的纖維化性肺疾病患者而言,目前僅有少量有限的有效治療可供選擇,需要進一步開展機制及轉化醫學等方面的研究。線粒體功能障礙引發纖維化反應的證據在IPF中尤為明顯,但是將線粒體質量控制與肺纖維化異常修復聯系起來的機制仍需進一步探索。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
中老年保健(2022年2期)2022-11-25 23:46:31
天天愛科學(2022年9期)2022-09-15 01:12:54
昆明醫科大學學報(2022年4期)2022-05-23 13:04:50
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
學苑創造·A版(2020年9期)2020-10-13 09:41:02
航空世界(2020年10期)2020-01-19 14:36:20
國際呼吸雜志(2019年21期)2019-11-25 09:52:20
天津醫科大學學報(2015年3期)2015-06-05 12:21:49
