固相微萃取技術在毛發藥物分析中的應用
2010-01-16 00:22:14孫英英
中國司法鑒定 2010年3期
關鍵詞:分析
孫英英,沈 敏
(1.蘇州大學基礎醫學與生物科學學院法醫學系,江蘇 蘇州215123;2.司法部司法鑒定科學技術研究所上海市法醫學重點實驗室,上海200063)
固相微萃取 (solid phase microextraction,SPME)技術是1990年由加拿大的Pawliszyn教授在固相萃取基礎上首先提出來的一種新的萃取分離技術,具有可與GC/MS、HPLC/MS等在線聯用、樣品量小、無需溶劑、重現性好等優點,可實現提取、濃縮、衍生化、分析的一體化。在法醫毒物學領域,SPME技術用于毛發分析的主要對象為濫用藥物。1998年,Koide[1]首先將SPME技術應用于毛發中苯丙胺類興奮劑分析,而后應用范圍不斷擴大,包括美沙酮、大麻、可卡因、氯胺酮、利多卡因等。對于SPME技術在毛發分析中的應用狀況分析有助于SPME技術發展及其應用潛力拓展,也促進實現毛發分析的快速、簡便、高效、靈敏。
1 SPME技術
1.1 SPME模式
SPME分為直接SPME(DI-SPME)和頂空SPME(HS-SPME)兩種模式。DI-SPME是將萃取纖維直接插入到待測樣品中,適于分析難揮發的待測物和較潔凈的樣品基質。當樣品基質成分復雜且含有生物大分子時,若采用DI-SPME則基質雜質易吸附在萃取纖維上,對色譜分析產生干擾或造成基線不穩,并致萃取纖維壽命減少;而HS-SPME是將萃取纖維置于待測樣品上方,利用待測物的易揮發性在頂空瓶上方將其吸附,因而可消除雜質干擾和基質影響,適于分析易揮發性待測物和復雜基質樣品。但若待測物質的沸點很高或有大分子干擾時,該方法耗時且靈敏度低,這是因為待測物沸點越高,越難揮發,因而頂空氣相中待測物濃度很低,不利于萃取。提高樣品溫度雖有助于樣品快速向頂空轉移,但是可能會造成頂空待測物與纖維涂層的吸附力降低。針對這一問題,Zhang等[2]設計一種中空的纖維膜,可保護熔融石英纖維,使待測物自由擴散通過而大分子無法通過,此膜可用于含有高分子干擾物的高沸點物質的測定。另外Zhang等[3]還設計了內冷式SPME,利用內置CO2冷卻裝置在加熱樣品的同時降低萃取纖維的溫度,從而提高萃取率。
1.2 SPME過程
SPME包括吸附和解吸兩個主要過程。當纖維暴露在樣品中時,涂層可以從液態/氣態基質中吸附待測物,然后將富集了待測物的纖維直接轉移到儀器中,通過一定的方式解吸后進行分離分析。圖1[4]顯示了HS-SPME分析頭發中苯丙胺類化合物的樣品水解、萃取、衍生化、分析的全過程。SPME采用熔融石英纖維,外面涂有適當的固定相,如聚丙烯酸酯(PA),聚二甲基硅氧烷(PDMS),聚二甲基硅氧烷/二乙烯基苯(PDMS/DVB)等。樣品中被分析物吸附于纖維涂層上,平衡后注入氣相色譜注射口進行熱解吸。
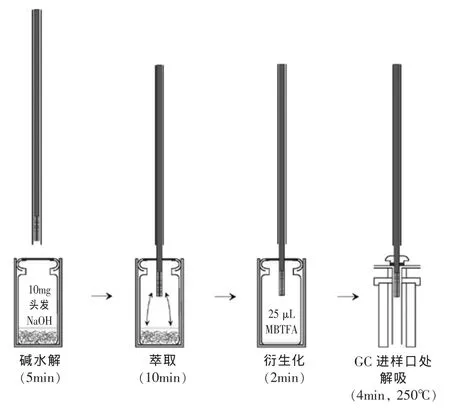
圖1 固相微萃取技術測定頭發中苯丙胺類化合物
在吸附和解吸附過程中,纖維涂層是至關重要的。涂層類型的選擇應綜合考慮待測物的分配系數、極性、沸點等因素,根據相似相溶原理,極性涂層對極性待測物具有較強的親和力,反之亦然,這就要求選擇的涂層性質應與待測物的性質相匹配。首先,涂層應對待測物有較強的富集能力,使待測物在纖維涂層中可以較快地擴散,快速達到分配平衡;其次,在解吸時待測物能迅速脫離纖維涂層,不會造成峰展寬。
目前商品化涂層有6種,按照極性大小可分為三類:一類為非極性涂層如PDMS,適用于分析非極性化合物;一類為極性涂層如PA和聚乙二醇(PEG),適用于分析極性化合物,其中PA適用于分析雜原子極性化合物,PEG適用于分析酚類化合物;一類為中等極性混合型涂層如聚乙二醇-二乙烯基苯 (CWDVB)。這三類涂層可滿足大多數生物樣品中待測物的萃取,但是仍存在很多缺陷如選擇性弱、使用壽命短、高溫下不穩定等。高效、高選擇性、長壽命的涂層是纖維發展的關鍵,科研工作者研究出很多的新型涂層如聚吡咯(PPY)涂層、纖維雙液相涂層、溶膠-凝膠等。PPY涂層可萃取極性或者離子型化合物,由于PPY涂層不僅具有高的萃取率,且在不同溶液中保持穩定,因此可滿足SPME與LC聯用的需要,擴大了分析范圍。溶膠-凝膠技術可選擇合適的有機組分結合到無機聚合物結構中,對極性和非極性化合物均有較強的萃取富集能力,分析時間較短,高溫下可正常使用,擴大了SPME技術的應用范圍。目前已經應用于生物樣品的分析[5],對酚類、胺類的回收率都高于一般萃取纖維。
此外,凡是影響吸附和解吸附的因素均會影響SPME的萃取效率。吸附效果可以從以下幾個方面優化:(1)萃取時間。萃取時間是待測物在纖維涂層和樣品間分配平衡所需要的時間,萃取初期待測物易到達纖維固定相富集,但隨著時間延長速度變慢,接近平衡狀態時萃取率不再隨時間發生變化,因此選擇萃取率不再改變之最短時間為吸咐時間。影響萃取速度的因素有攪拌和溫度。攪拌可以加快待測物在涂層和樣品間的分配,很多實驗中發現攪拌能明顯提高萃取率,縮短平衡時間。攪拌的方式有磁力轉子攪拌、高速勻漿和超聲,到達平衡的時間依次遞減,其中超聲效果最好,但由于磁力轉子攪拌所用設備簡單而應用廣泛。萃取溫度對SPME的影響具有雙重作用[6]:一方面加熱可以加速樣品分子運動的速度,有利于分析物在基質中擴散,縮短平衡時間;另一方面過高的溫度會降低石英纖維固定相對組分的吸附能力,因此選擇合適的溫度十分重要。(2)溶液pH值。調整pH使溶液中大部分待測物處于等電點或者接近等電點,從而處于分子狀態易被固定相吸附,因此酸性物質需要在酸性環境中萃取,堿性物質需要在堿性環境中萃取。(3)溶液中鹽濃度。適量K2CO3等鹽類,可增強離子強度降低極性有機化合物在水中的溶解度而易被石英纖維固定相萃取,但并不是對所有待測物均有效,有時鹽類的加入也能提高被測物的溶解度。
1.3 SPME與色譜法的聯用技術
SPME萃取完成后往往將萃取纖維直接插入進樣口或接口進行解吸,根據聯用技術的不同,有兩種解吸過程。與GC聯用時萃取頭纖維直接插入GC進樣口中進行熱解吸,使待測物從纖維上脫吸附進入色譜柱中進行分析;與HPLC聯用時萃取頭纖維直接放入SPME和HPLC的接口處,利用合適的溶劑進行溶解后隨流動相進入色譜柱內分析。
1.3.1 SPME-GC聯用技術
SPME-GC聯用技術已相對成熟,廣泛應用于毛發中濫用藥物的分析,主要分析揮發性和半揮發性有機物,時間一般在30min內,檢出限(LOD)達ng/mgpg/mg級水平,線性范圍較寬。GC檢測器主要應用MSD和NPD。Oliveira等[7]應用HS-SPME和GC-MS聯用測定犬毛發中揮發性化合物,該方法在90℃用65μm PDMS萃取18min后在GC進樣口240℃解吸1min,成功分離檢測到犬毛發中250種揮發性化合物并確定了幾種用于疾病診斷的生物標志物。Koide等[1]應用HS-SPME和GC-NPD聯用檢測頭發中未經衍生化的苯丙胺和甲基苯丙胺。
運用SPME-GC時應注意在不破壞所選涂層的前提下,應使用最高溫度及最小直徑的進樣器插口,這樣可使解吸時間縮短且可排除殘留物的影響;插入纖維后應盡可能快地將纖維暴露出來以減少吸附物質在針內解吸附而導致分裂峰產生;最后還要注意萃取頭在氣化室中的位置,氣化室頂部溫度要低于中部和下部,因此萃取頭應盡量伸出,使解吸完全。
1.3.2 SPME-HPLC聯用技術
適合HPLC分析的化合物范圍較GC寬泛得多。Chen等[8]報道了SPME與HPLC聯用,分析熱不穩定以及低揮發性或不揮發性的強極性化合物。HPLC中選用適當的溶劑解吸,速度較快可克服SPME-GC中出現的延遲現象。SPME-HPLC在其它生物檢材中有機物的分析已有應用[9],但在毛發中濫用藥物的分析尚未見報道,可能是由于以下原因:SPME-HPLC的接口技術是聯用技術的關鍵所在,此接口裝置具特殊要求;HPLC/MS無專門的圖譜庫,對于未知物分析可因圖譜庫功能的不完善而產生錯誤結果;溶劑洗脫時可產生顯著的柱外擴散效應,這些因素限制了該技術在毛發分析方面的應用。新近研制成功的自動采樣SPME-LC/MS技術可有效解決SPME-HPLC聯用產生的柱外擴散,縮短萃取時間,提高方法精密度。
1.4 SPME的衍生化技術
SPME用于極性有機化合物的分離分析常涉及衍生化問題。SPME衍生化主要有三種類型:(1)直接衍生化即將衍生化試劑直接加到樣品溶液中,萃取纖維從頂空或溶液中萃取衍生化的化合物,然后送到進樣口。(2)在SPME的萃取纖維上衍生化即萃取被測物后,將萃取頭置于衍生化試劑的氣相或直接置于衍生化溶液中衍生化。(3)在GC進樣口衍生化即衍生化在GC的進樣口與熱解吸同時完成。
為了適應大范圍篩選的需要[10],所選擇的衍生化試劑應盡可能對濫用藥物及其代謝物的所有活性基團進行衍生化以適于氣相色譜分析。常用的衍生化試劑有酰化試劑、硅烷化試劑以及鹵代酰化試劑。其中硅烷化試劑在毛發濫用藥物的分析中最為常用,其衍生化后無需揮干試劑可直接進樣。鹵代酰化試劑衍生化后有較好的色譜行為,碎片離子特征明顯,但是對于像大麻酸這類分子量較大的物質,衍生化后分子量大不利于質譜檢測。當使用以上兩類作為衍生化試劑時衍生化反應均需嚴格控制無水。另外據Solans[11]報道硅烷化(TMS)和多氟酰化(TFA)的雙衍生化方法是一種良好的選擇。N-甲基三甲硅基三氟乙酰胺(MSTFA)等硅烷化試劑可選擇性地與含有羥基、羧基、酚基化合物及代謝物如嗎啡、單乙酰嗎啡、苯甲酰芽子堿反應形成O-TMS衍生化物;MSTFA則與含有氮基的伯胺、仲胺如苯丙胺類反應形成N-TFA衍生化物。雙衍生化的次序為TMS化在前,TFA化在后。
2 SPME在毛發中濫用藥物檢測的應用
2.1 苯丙胺類興奮劑
Koide等[1]用SPME和GC/NPD聯用技術快速檢測頭發中甲基苯丙胺(MA)及其主要代謝產物苯丙胺(AP)。將頭發置于NaOH溶液中水解,用PDMS吸附萃取,發現當溫度升高時MA和AP的萃取率增加并在55℃時獲得最大值,此后隨著溫度的升高兩者萃取率大幅度下降,表明萃取溫度在55℃時可獲得最大萃取率。另外隨著萃取時間的延長MA和AP的萃取率不斷升高,但萃取時間超過15min后,待測物與內標比例不再發生改變,為了節省分析時間且獲得最佳結果,最終選擇的萃取時間和溫度分別為 20min和55℃。解吸時觀察220℃時0.1~1min內5個不同時間點的解吸效率,發現不到0.5minMA和AP的解吸率分別大于99%和97%,表明MA和AP的解吸是瞬間完成的,可能是因為加熱至220℃所產生的熱量遠遠高于待測物與纖維之間的吸附作用力。為了使待測物完全解吸并延長萃取頭壽命,采用的解吸條件為250℃,3min。在此條件下測定頭發中AP和MA的線性范圍分別為 0.4~15ng/mg和 4~160ng/mg(R>0.998);LOD分別為0.1ng/mg和0.4ng/mg,且GC上無共存雜質峰出現,表明該方法靈敏度高且專屬性強。
當使用GC/MS分析時為了獲得專屬的質譜圖,往往將AP進行全氟衍生化,但是此類衍生化試劑反應時需要保證無水條件,而大多數反應是在水溶液中進行的,因此可采用n-丙基氯甲酸酯、五氟芐基溴、七氟丁酰氯(HFB-Cl)等作為衍生化試劑。Liu等[12]采用HS-SPME和GC/MS聯用對頭發中苯丙胺衍生物進行定性定量分析。將HFB-Cl加入到水解液中進行衍生化,然后將PDMS放入頂空瓶中于60℃萃取20min后直接插入GC進樣口250℃解吸3min,發現經HFB-Cl衍生化后AP和MA的色譜行為得到很大改善,AP和MA在0.1~100ng/mg范圍內線性相關(R>0.999),所獲得的質譜專屬性良好且靈敏度是傳統方法的五倍。Musshoff等[13]也采用HS-SPME和GCMS聯用分析頭發中苯丙胺衍生化產物,但應用的衍生化試劑MBTFA對隨后的分析步驟和纖維都有一定的影響,因而不適合作常規分析;且因分析過程中需打開頂空小瓶、添加衍生化試劑而不適于自動分析。Nishida等[14]應用烷基氯仿進行衍生化時注意到苯丙胺類及其代謝產物具有揮發性而需密封頂空小瓶。考慮到密封材料可能對HS-SPME產生影響,因而分別對橡膠塞、具有鋁膜的橡膠塞和硅質塞進行分析,發現橡膠塞對衍生化產物具有吸附作用,測得的回收率較低,鋁膜的加入可在一定程度上改善回收率但效果不如硅質塞好。頭發在1mol/L NaOH溶液水解20min,LOD為0.02ng/mg(MA)和0.05ng/mg(AP),絕對回收率范圍為2.8%~17.5%。
對比兩種方法發現利用HS-SPME和GC/MS聯用檢測頭發中AP和MA具有一定優勢,線性范圍寬,靈敏度較高,LOD低,且衍生化后可獲得特征質譜圖。
2.2 可卡因及其代謝產物苯甲酰芽子堿、古柯乙烯
Toledo等[15]建立一種SPME與GC-MS聯用方法對頭發中可卡因、苯甲酰芽子堿、古柯乙烯同時進行定性定量分析。大多數頭發中使藥物游離采用的方法為堿性水解,而酯類化合物長時間放于堿性化學環境中易發生分解并且影響萃取頭壽命,該文比較了甲醇浸潤和堿性水解兩種游離方法,發現甲醇可游離頭發中大部分待測物而堿性水解后獲得的物質大部分是可卡因及其代謝產物的水解產物,表明可卡因及其代謝產物苯甲酰芽子堿、古柯乙烯等酯類化合物應在甲醇中提取以避免在強堿環境中發生水解。可卡因及其代謝物游離后加入催化劑乙腈、吡啶進行烷基化,采用的衍生化試劑是丁基氯仿。將兩種衍生化方法加以比較:一種是甲醇游離待測物后吹干再加入乙腈、吡啶和衍生化試劑丁基氯仿進行衍生化;另一種是參照Hall[16]的方法,直接向帶有頭發的甲醇溶液中加入乙腈、水、正己烷等催化劑和衍生化試劑己基氯仿進行衍生化,發現前者萃取效果良好,低濃度時三者回收率在74.6%~88.4%,中、高濃度的三者回收率接近100%,利用GC-MS分析可卡因和苯甲酰芽子堿的LOD和定量限(LOQ)分別為0.1ng/mg和0.5ng/mg,古柯乙烯的LOD和LOQ均為0.1ng/mg,但是后者方法的回收率很低,可能是由于頭發基質復雜,將PDMS直接放入基質中萃取衍生化物質時頭發中蛋白質在纖維上吸附且可容易達到分配平衡,從而影響待測物的萃取及分配平衡;另外因基質影響反應進行而使衍生化效率降低從而影響萃取率。萃取完后直接將PDMS放入250℃的GC進樣口熱解吸20min。MS在 SIM方式下進行定性定量分析,在0.1~50ng/mg范圍內線性相關(R>0.98);日間、日內精密度良好。
除了甲醇提取可卡因外,也有作者采用酸性提取。Gentili等[17]利用HS-SPME分析頭發中濫用藥物時針對可卡因的特殊性質,對其進行專門的條件優化,在酸性環境中利用PDMS萃取頭發中可卡因,并考察不同HCl濃度對回收率的影響。向含有1mol/L HCl和0.1mol/L HCl的頭發基質中加入80mg K2CO3時,雖然所測得的pH相差不大分別為10.3和11,但是可卡因的回收率卻有很大的不同,隨著HCl濃度的增加,可卡因的回收率也升高甚至超過60%,說明1mol/L HCl并未引起頭發基質可卡因的分解,反而有利于可卡因的萃取。溶液中加入K2CO3有利于可卡因的快速萃取,萃取時間僅需5min,表明K2CO3的加入使溶液中離子強度增加從而使可卡因的溶解度降低,有利于PDMS吸附。
同時攝入可卡因和乙醇可產生毒性很強的代謝產物古柯乙烯,在臨床和毒物分析中對其分析也變得很重要。Bermejo等[18]采用酶水解頭發,用緩沖溶液調節pH后加入NaCl,用100μm PDMS萃取25min,然后直接插入GC進樣口于250℃解吸5 min。可卡因和古柯乙烯的LOD分別為0.08ng/mg和0.02ng/mg,測得的靈敏度和相關系數比Gentili[17]和Toledo[15]測得的要高,主要因為酶水解較酸水解更完全,獲得的回收率高且萃取物更為干凈無干擾;100μm PDMS對極性化合物可卡因和古柯乙烯的萃取率較高且平衡時間較短。
2.3 大麻酚類
大麻的主要成分是大麻酚類,其中生物檢材中經常檢測的大麻酚類分別為四氫大麻酚(THC)、大麻二酚(CBD)和大麻酚(CBN),其代謝物四氫大麻酸(THC-COOH)的脂溶性很強且較難與毛發基質結合。Musshoff等[19]首次將大麻酚類衍生化后用HS-SPME萃取,GC-MS分析。優化條件測得三者在0.1~20ng/mg范圍內呈線性相關(R>0.998),LOD為0.05ng/mg(THC)、0.08ng/mg(CBD)和0.14ng/mg(CBN),絕對回收率為0.3%~7.5%,雖然絕對回收率不高,但相對回收率遠高于液液萃取,主要是因為前者纖維針直接插入GC進樣口將所吸附的待測物全部解吸并進行分析而后者僅為部分萃取物。
常用于大麻酚類的衍生化試劑為BSTFA和五氟丙酸酐(PFPA),較常采用BSTFA衍生化主要是因為BSTFA衍生化無需吹干即可進樣,可明顯改善峰形,提高靈敏度并且可以避免PFPA的酸性對纖維的損害,但是在毛發分析中BSTFA衍生化有一缺陷,即在圖譜中可觀察到THC處有雜質干擾峰出現。萃取時高溫有助于待測物由液相向氣相中擴散,因此隨著溫度升高其相對萃取率也不斷升高,分別在 120℃(THC)和70~80℃(CBD,CBN)獲得最大值。鑒于以下兩方面原因最終選擇的溫度為90℃:一方面應綜合考慮溫度對所有待測物的影響而不僅僅考慮對某種物質的影響,酚類在90℃獲得的相對回收率范圍為40% ~80%;另一方面選擇的萃取溫度應低于鹽的沸點以避免氣相中揮發鹽的水分在頂空瓶膜口處由于溫度的降低而凝結,從而污染纖維。熱解吸時在200~270℃測定0.5~10min內的解吸率,發現相對回收率隨著溫度的升高而增加,在250℃時獲得最大值,此后的高溫會造成相對回收率的下降,且高溫對PDMA的壽命有不利影響,因此最佳解吸溫度為250℃,解吸5min。
為了進一步提高THC、CBD和CBN檢測方法的靈敏度,Nadulski等[20]采用丙酮萃取待測物后揮干,在殘留物中加入 BSTFA進行衍生化后再用 100μm PDMS頂空萃取。此方法的特別之處在于萃取前先用液液萃取的方法將待測物萃取出來,然后揮干有機溶劑再在衍生化試劑中頂空萃取,LOD大大降低,為0.01~0.02ng/mg。得到該結果有以下幾方面的原因:首先,水溶液中的大麻酚類物質很難被PDMS吸附,而將其用丙酮萃取后在衍生化試劑中易于吸附;其次,揮干丙酮后使待測物濃縮更易于PDMS吸附;再者,在衍生化試劑中進行HS-SPME時可使用較高的萃取溫度,而高溫有助于提高萃取率。實驗還對液液萃取的有機溶劑、衍生化試劑和用量、萃取纖維類型與厚度、萃取時間和溫度等條件進行優化,最終獲得樣品中THC、CBD和CBN總回收率分別為8.9%、59%和6.4%,三者有較大差異是因為在HS-SPME操作中標準物質直接添加至丙酮中而不是頭發樣品中,此步測得絕對回收率分別為29%、66%和14%。另外CBD回收率高可能是因為其結構有兩個羥基均可進行衍生化,因而衍生化效率比較高,衍生化產物易于被PDMS吸附。
2.4 美沙酮
Lucas[21]利用SPME-GC/MS同時測定頭發中美沙酮及其代謝產物吡咯烷(EDDP),先用蛋白酶E水解頭發12h后在37℃用PDMS萃取30min,然后直接放在GC進樣口于250℃解吸5min。結果:在1.0~50ng/mg范圍內線性相關(R>0.995);在3.0ng/mg和30.0ng/mg時美沙酮和 EDDP的 RSD分別小于 13.30%和8.94%;二者回收率均接近100%。在此條件下分析183個美沙酮攝入者的陽性頭發樣本,考察頭發顏色對美沙酮含量的影響,當攝入量均為80mg/day時黑色和棕黑色頭發中美沙酮的含量分別為25.12ng/mg和 31.31ng/mg,EDDP的含量分別為 2.97 ng/mg和5.07ng/mg,可見頭發顏色對美沙酮含量的影響不明顯,這可能是因為頭發中藥物主要是和黑色素結合而黑色和深棕色之間顏色差異較小的緣故。
Sporkert等[22]也應用HS-SPME-GC/MS同時檢測頭發中美沙酮及其兩種代謝產物EDDP和吡咯啉(EMDP),對于頭發中EMDP的檢測尚屬首例。首先,對纖維涂層的類型進行優化,同時應用65μmPDMS/ DVB和 85μmPA萃取頭發中待測物,發現應用PDMS/DVB萃取時EDDP獲得的回收率要高出25%,而用PA萃取時美沙酮的回收率要高出20%,由于頭發中EDDP的濃度較低,為了保證EDDP測定的靈敏度,采用65μmPDMS/DVB作為萃取纖維;其次,對水解溶液中NaOH的濃度和鹽的種類進行優化,發現隨著NaOH濃度的升高,美沙酮的萃取率逐漸降低而EDDP的回收率基本保持不變,加入0.5gNaCl比Na2SO4測得待測物的回收率要高,因而選擇水解液的組成為1mol/L NaOH+0.5gNaCl;再者,對萃取時間和溫度進行條件優化,測定60~120℃下5~50min內待測物的回收率,發現隨著溫度升高待測物的回收率均增加,但在100~110℃時美沙酮的回收率驟增而EDDP的回收率增加緩慢,30min后由于待測物降解兩者的回收率均下降,因此采用110℃萃取20min;最后,對樣品加入量進行條件優化,在堿性介質中隨著頭發基質的增加美沙酮的回收率下降,當加入全部頭發基質時美沙酮的回收率僅相當于未加入頭發基質的10%,EDDP的回收率先增加但超過10mg時也會降低,這主要是因為親脂性藥物富集于溶液表面,有利于低揮發性化合物的萃取,而加入頭發基質時由于基質產生其它親脂性物質的取代作用,造成溶液表面藥物濃度的降低,適宜的樣品量為10mg。
樣品在1mol/L NaOH和0.5gNaCl溶液中水解后,用65μmPDMS/DVB在110℃萃取20min然后直接解吸,所測得的LOD分別為0.03ng/mg(美沙酮)和0.05ng/mg(EDDP、EMDP),LOQ分別為0.10ng/mg(美沙酮)和0.16 ng/mg(EDDP、EMDP),絕對回收率分別為10.5%~11.2%(美沙酮)、11.0%~14.5%(EDDP)和15.9%~17.4%(EMDP)。
2.5 利多卡因
利多卡因是一種常見的麻醉劑,近幾年發現利多卡因致死案件逐漸增多,毛發利多卡因分析發現其很容易進入并儲存在頭發中。Sporkert等[23]利用HS-SPME分析49個案件樣品,比較了頭發和血液中利多卡因的含量,其中32個案件在頭發中測得利多卡因,含量范圍為0.4~300ng/mg,一例案件甚至達到675 ng/mg,而血液中可檢測到利多卡因的案件僅為11例。
頭發中利多卡因的測定使用65μm CW-DVB纖維吸附水解液中待測物,在分析過程中針對水解液組成、吸附時間和溫度進行優化,最終采用4%NaOH和0.5g Na2SO4作為水解液。對Na2SO4在NaOH分解頭發過程中的影響進行分析,發現Na2SO4加入順序對萃取無顯著性影響,說明Na2SO4并不影響頭發中藥物的游離。在60~70℃吸附15min,萃取完成后在250℃解吸5min,測得的絕對回收率為1.8%~2.4%,檢測的背景干擾小,LOD和LOQ分別為0.1ng/mg和0.4ng/mg。案例分析發現利多卡因主要摻雜在可卡因制劑中,有時也作為可卡因-海洛因制劑的摻雜物,并且可在大多數藥物濫用者的頭發中檢出。
2.6 尼古丁
分析頭發中尼古丁及其代謝產物可鐵寧可以區分吸煙者、被動吸煙者和非吸煙者。Sporkert等[24]利用SPME分析頭發中尼古丁,測得尼古丁的含量范圍為0.4~63.5ng/mg,在兩個相關案例中利用PA纖維萃取15min測得尼古丁含量分別為135ng/mg和334ng/mg,雖然可鐵寧在NaOH溶液中性質穩定,但是在吸煙者的頭發樣本或不含有頭發的標準溶液中均無法檢測到可鐵寧,可能是因為可鐵寧的親水性強而未能在纖維上富集。Sporkert等總結了很多案例發現應用HSSPME測定叔胺類物質時無法檢測到N-去甲基代謝產物,這可能是因為去甲基代謝產物的極性大,導致GC/MS檢測的靈敏度很低。
Gentili等[17]建立HS-SPME-GC/MS快速篩選方法可同時分析毛發中濫用藥物,包括可卡因、氯胺酮、美沙酮、苯丙胺、甲基苯丙胺、MDA、MDMA、MDE、MBDB等,測得各種化合物的LOD范圍為0.35~1.61ng/mg,相對回收率范圍為96.00%~105.50%。同時應用此方法分析183個頭發樣本中各種物質發現可卡因所占比例很高,另外還發現有些頭發樣本在檢測到可卡因的同時也檢測到利多卡因。Musshoff等[4]也提出SPME作為一種新技術在頭發分析中有著廣泛的發展前景,并指出在應用SPME分析時需要注意條件優化的重要性,包括萃取時間和溫度、衍生化時間、衍生化試劑的種類和用量、解吸時間和溫度等。
3 展望
目前已有多篇關于SPME在毛發藥物分析應用的研究綜述[24-25],但該類研究仍然處于起步階段。由于毛發基質復雜、毛發中待測物含量低且大部分待測物揮發性較弱,導致SPME在頭發分析中的應用具有一定的局限性。
SPME法的基質和待測物的適用范圍拓展在于SPME技術自身發展,未來SPME將面臨的挑戰:一是要增加萃取纖維涂層的種類;二是要提高纖維涂層的熱穩定和耐侵蝕性;三是提高纖維涂層的選擇性和識別能力;四是完善SPME與HPLC-MS的聯用技術,五是SPME應用研究的積累。隨著科學技術的發展,有理由相信,SPME法將在毛發藥物分析領域有著更為廣闊的應用前景。
[1]Koide I,Noguchi O,Okada K,et al.Determination of amphetamine and methamphetamine in human hair by headspace solid-phase microextraction and gas chromatography with nitrogen–phosphorus detection[J].J Chromatogr.B,1998,707(1-2):99.
[2]Zhang ZY,Poerschmann J,Pawliszyn J.Direct solid microextraction of complex aqueous samples with hollow fiber membrane protection[J].Anal Commun,1996,33(7):219.
[3]Zhang ZY,Pawliszyn J.Quantitative Extraction using an internally cooled solid phase microextraction device[J].Anal. Chem.,1995,67(1):34243.
[4]Musshoff F,Madea B.New trends in hair analysis and scientific demands on validation and technical notes[J].Forensic Sci.Int.,2007,165(2-3):204.
[5]Wang D,Xing J,Peng J,et al.Novel benzo-15-crown-5-solgel coating for solid-phase microextraction[J].J Chromatogr.A,2003,1005(1-2):1.
[6]傅彥斌.固相微萃取分析條件的優化[J].Arid Cnuironmentdal Monitoring[J].2006,20(1):49.
[7]Oliveira LS,Rodrigues F,Oliveira FS,et al.Headspace solid phase microextraction-gas chromatography– mass spectrometry combined to chemometric analysis for volatile organic compounds determination in canine hair:A new tool to detect dog contamination by visceral leishmaniasis[J].J Chromatogr.B,2008,875(2):392.
[8]Chen J,Pawliszyn J.Solid phase microextraction coupled to high performance liquid chromatography[J].Anal.Chem,1995,67(15):2530.
[9]沈敏,向平.毒鼠強中毒的固相微萃取和GC/NPD快速測定[J].中國藥學,2000,35(5):341.
[10]沈敏.體內濫用藥物分析.北京:法律出版社,2003:48.
[11]Solans A,Carnicero M,Delatorre,et al.Comprehensive Screening Procedure for Detection of Stimulants,Narcotics,Adrenergic Drugs and Their Metabolites in Human Urine[J]. Anal Toxicol.1995,19(2):104.
[12]Liu JT,Hara K,Kashimura S,et al.New method of derivatization and headspace solid-phase microextraction for gas chromatographic–mass spectrometric analysis of amphetamines in hair[J].J Chromatgr.B,2001,758(1):95.
[13]Musshoff F,Junker HP,Lachenmeier DW,et al.Fully automated determination of amphetamines and synthetic designer drugs in hair samples using headspace solid-phase microextraction and gas chromatography-massspectrometry[J]. J ChromatogrSci,2002,40(6):359.
[14]Nishida M,Yashiki M,Namera A,et al.Single hair analysis of methamphetamine and amphetamine by solid phase microextraction coupled with in matrix derivatization[J].J Chromatgr.B,2006,842(2):106.
[15]Toledo FCP,Yonamine M,Moraes Moreau RL,et al. Determination of cocaine,benzoylecgonine and cocaethylene in human hair by solid-phase microextraction and gas chromatography–mass spectrometry[J].J Chromatogr.B,2003,798(2):361.
[16]Hall BJ,Parikh AR,Brodbelt JS.Aqueous phase hexylchloroformate derivatization and solid phase microextraction:determination of benzoylecgonine in urine by gas chromatography-quadrupole ion trap mass spectrometry[J].Forensic Sci.,1999,44(3):527.
[17]Gentili S,Cornetta M,Macchia T.Rapid screening procedure based on headspace solid-phase microextraction and gas chromatography–mass spectrometry for the detection of many recreational drugs in hair[J].J Chromatogr.B,2004,801(2):289.
[18]Bermejo AM,López P,álvarez I,et al.Solid-phase microextraction for the determination of cocaine and cocaethylene in human hair by gas chromatography–mass spectrometry[J].Forensic Sci.,2006,156(1):2.
[19]Musshoff F,Junker HP,Lachenmeier DW,et al.Fully Automated Determination of Cannabinoids in Hair using Headspace Solid-Phase Microextraction and Gas Chromatography–Mass Spectrometry[J].J Anal.Toxicol.2002,26(8):554.
[20]Nadulski T,Pragst F.Simple and sensitive determination of△-9-tetrahydrocannabinol,cannabidiol and cannabinol in hair by combined silylation,headspace solid phase microextraction and gas chromatography–mass spectrometry[J].J Chromatogr.B,2007,846(1-2):78.
[21]Lucas ACS,Bermejo AM,Tabernero MJ,et al.Use of solid-phase microextraction(SPME)for the determination of methadone and EDDP in human hair by GC–MS[J].Forensic Sci.Int.,2000,107(1-3):225.
[22]Sporkert F,Pragst F.Determination of methadone and its metabolites EDDP and EMDP in human hair by headspace solid-phase microextraction and gas chromatography–mass spectrometry[J].J Chromatogr.B,2000,746(2):255.
[23]Sporkert F,Pragst F.Determination of Lidocaine in Hair of Drug Fatalities by Headspace Solid-Phase Microextraction[J]. Anal.Toxicol.,2000,24(5):316.
[24]Sporkert F,Pragst F.Use of headspace solid-phase microextraction(HS-SPME)in hair analysis for organic compounds [J].Forensic Sci.Int.,2000,107(1-3):129.
[25]劉曉茜,沈敏.固相微萃取在司法化學鑒定中應用的進展[J].中國司法鑒定,2004,(2):15.
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
