全自動固相萃取分子篩脫水氣質(zhì)聯(lián)用法測定水中有機磷農(nóng)藥殘留
2015-02-25 07:11:47秦明友上海市金山區(qū)環(huán)境監(jiān)測站上海200540
西部皮革 2015年8期
關(guān)鍵詞:氣相色譜
秦明友(上海市金山區(qū)環(huán)境監(jiān)測站,上海200540)
全自動固相萃取分子篩脫水氣質(zhì)聯(lián)用法測定水中有機磷農(nóng)藥殘留
秦明友
(上海市金山區(qū)環(huán)境監(jiān)測站,上海200540)
摘要:建立了氣相色譜質(zhì)譜聯(lián)用法測定水中有機磷農(nóng)藥殘留的方法,樣品用全自動固相萃取分子篩脫水進行預(yù)處理,對固相萃取盤的類型進行了比選,試驗了萃取盤的穿透性,優(yōu)化了萃取條件,用丙酮和甲基叔丁基醚(MTBE)作萃取劑,用DVB萃取盤進行萃取。方法相關(guān)系數(shù)(r2)為0.9930~0.9991,檢出限為0.000006~0.000018 mg/L,方法用于飲用水、工業(yè)廢水和生污水中有機磷農(nóng)藥的測定,對高、中、低濃度的樣品進行加標回收測定(n=6),精密度為2.7%~16.7%,平均加標回收率為71.7%~101.2%。方法具有良好精密度、準確度和靈敏度,前處理方法簡單快速,環(huán)境友好。
關(guān)鍵詞院有機磷農(nóng)藥;分子篩脫水;自動固相萃取;氣相色譜/質(zhì)譜;水
有機磷農(nóng)藥是一類高效、廣譜的殺蟲劑,具有成本低、藥效高、品種多、選擇性好、防治范圍廣等特點,但都具有高毒或劇毒,對人體有極大的危害。研究表明,在日常施用農(nóng)藥的過程中,只有1%作用于靶標生物,其余的殘留于土壤或通過徑流進入水環(huán)境,并且有機磷農(nóng)藥有較長的殘存期,嚴重污染水環(huán)境,特別是集中式生活飲用水。因此,分析檢測地表水中有機磷農(nóng)藥殘留量具有較大的現(xiàn)實意義。目前,有機磷農(nóng)藥殘留主要采用氣相色譜法[1-5]、液相色譜法[6-9]、氣相色譜質(zhì)譜法[10-12]和液相色譜質(zhì)譜法[13],因氣相色譜良好的分離功能和質(zhì)譜良好的定性能力而廣泛應(yīng)用于有機磷農(nóng)殘的測定,但如果制備的樣品中有水,要損傷質(zhì)譜儀和降低質(zhì)譜儀的靈敏度,另外有水存在時,水與有機溶劑不互溶,當定容到1 mL時有機溶劑中目標化物的的濃度就不準確,因此在樣品處理過程中要除去萃取液中的水。傳統(tǒng)的脫水方法用Na2SO4脫水,該方法要將Na2SO4在400℃下烘烤4 h,脫水前要用溶劑潤洗,脫水完后還要用有機溶劑沖洗,方法操作繁鎖,消耗試劑Na2SO4,同時也消耗了更多的有機溶劑,污染了環(huán)境,增加了氮吹濃縮時間,同時也增多了樣品轉(zhuǎn)移次數(shù),從而降低了回收率。本實驗選用分子篩脫水,該分子篩用聚四氟乙烯作原料,做成一定大小的微孔,再用一些親有機物而疏水的材料涂于微孔之間,使有機物能通過微孔而水不能通過。方法操作簡單,樣品轉(zhuǎn)移次數(shù)少,不增加有機溶劑的量,脫水效果好,是一種環(huán)境友好型的前處理脫水方法。
1 實驗部分
1.1儀器與試劑
美國Agilent 7890A/5975C GC/MS聯(lián)用儀;色譜柱DB-624 30 m×0.25 mm×0.14 um; Horizontechnology SPE-DEX4790自動固相萃取儀;LABCONCO RapidVap N2/48自動氮吹儀;Millipore synergy純水機;Horizon 47 mm C18、DVB、HLB萃取盤;分子篩脫水杯。
有機磷農(nóng)藥:AccuStandard 100 ug/mL 1 mL(敵敵畏(dichlorvos),內(nèi)吸噒-S(demeton-s),樂果(dimethoate),內(nèi)吸磷-O (demeton-o),甲基對硫磷(parathion-methyl),馬拉硫磷(malathion),對硫磷(parathion) );菲D10(Ph-D10,內(nèi)標物):Dr. Ehrenstorfer GmbH25mg;PCB209(十氯聯(lián)苯,替代物):Dr. Ehrenstorfer GmbH 20 mg;超純水:電阻率為18.25 mΩ·cm甲醇:DUKSAN色譜純;丙酮J&K Superpure;MTBE(甲基叔丁基醚):CNW色譜純;高純氦氣(>99.999%);高純氮氣(>99.999%)。
1.2儀器條件
1.2.1色譜條件
50℃保持0.5 min,25℃/min到100℃,保持2 min,60 ℃/min到260℃,保持7 min;He為恒流,流速為1.0 mL/min進樣口溫度250℃,不分流模式,總流量14 mL/min,隔墊吹掃流量3 mL/min;MS接口溫度250℃。
1.2.2質(zhì)譜條件
EI離子源,70 EV,離子源溫度280℃,四級桿溫度150℃,增益系數(shù)5,溶劑延遲時間為8.0 min,數(shù)據(jù)采集模式選擇離子模式(SIM)。
1.2.3自動固相萃取條件
萃取盤活化:丙酮浸泡90 min,干吹90 min;甲醇浸泡90 min,干吹90 min;水浸泡90 min,干吹90 min。
萃取盤干吹:8 min。
萃取盤洗脫:丙酮浸泡90 min,干吹60 min;MTBE浸泡90 min,干吹60 min;MTBE浸泡90 min,干吹120 min;MTBE浸泡90 min,干吹120 min。
1.2.4自動氮吹儀條件
氮吹儀振蕩速度:最大值的60%,氮吹溫度:30℃。
1.3實驗方法
1.3.1樣品的制備
取500 mL經(jīng)0.45 um微孔濾膜過濾后水樣(清潔地表水不用過濾),加入一定量的替代物PCB209,裝入自動固相萃取儀的進樣瓶,按優(yōu)化的萃取條件進行萃取,萃取液(大約5 mL左右)轉(zhuǎn)移至脫水杯中進行脫水,用自動氮吹儀的氮吹杯(帶有定量刻度線)收集脫水后的萃取液,用1 mL甲基叔丁基醚洗脫水杯兩次,甲基叔丁基醚一并收集在氮吹杯中。將氮吹杯放入自動氮吹儀,按優(yōu)化的氮吹條件進行氮吹,當濃縮至0.8 mL左右時,停止氮吹,加入內(nèi)標物菲D10,用甲基叔丁基醚定容至1.00 mL,轉(zhuǎn)移至1.5 mL進樣瓶中備用。
1.3.2樣品的測定
將制備好的樣品用GC/MS選擇離子模式進分析,保留時間和特征離子定性,菲D10作內(nèi)標用內(nèi)標法進行定量。
2 結(jié)果與討論
2.1色譜圖
目標化合物的色譜圖見圖1,目標化合物保留時間和選擇離子見表1。
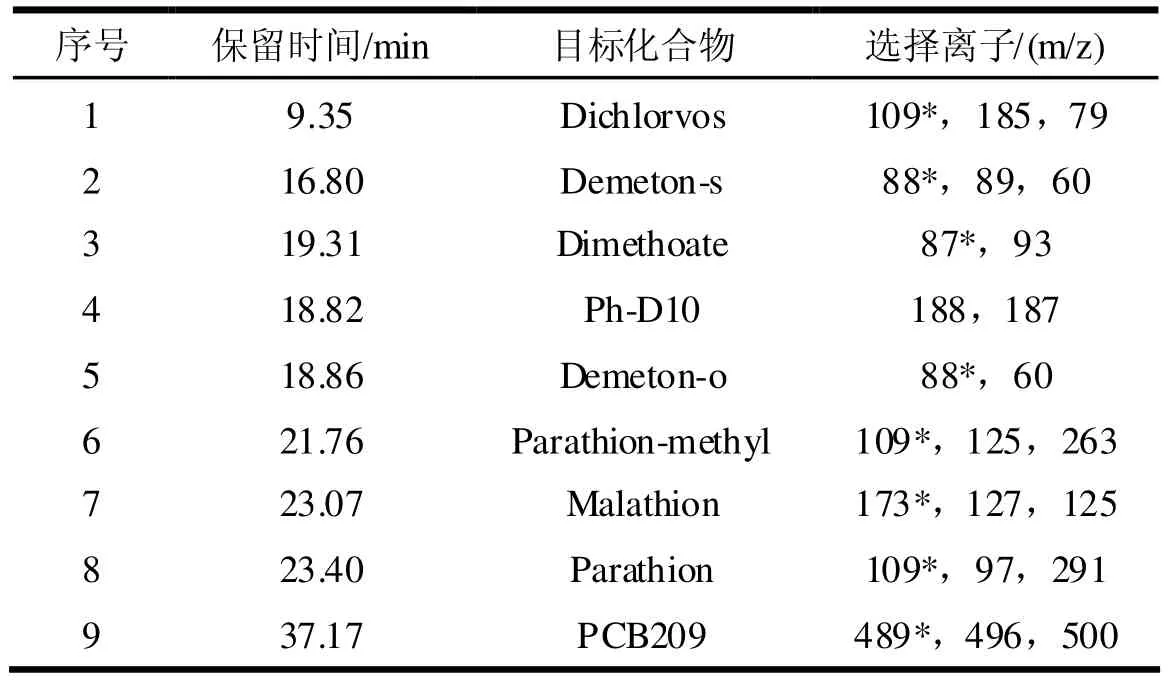
表1 保留時間及選擇離子

圖1 目標化合物的色譜圖
2.2條件的優(yōu)化
2.2.1色譜質(zhì)譜
離子源溫度升高和增益系數(shù)的增大可使質(zhì)譜響應(yīng)顯著增大,有機磷農(nóng)藥的質(zhì)譜響應(yīng)信號較弱,為提高方法靈敏度,將離子源溫度設(shè)定為280℃,增益系設(shè)置為5。
2.2.2樣品處理
(1)萃取介質(zhì)的選擇
固相萃取中固定相的性質(zhì)是影響萃取效率的重要因素,按實驗步驟對分別用固定相為C18、HLB和DVB的萃取盤對0.00001、0.0001和0.001 mg/L的有機磷農(nóng)藥溶液分別進行三次平行測定,其三次測定的平均回收率見表2,從表中分析可知,DVB萃取盤的萃取效果要明顯優(yōu)于C18和HLB萃取盤,
本實驗選用DVB萃取盤進行萃取實驗。
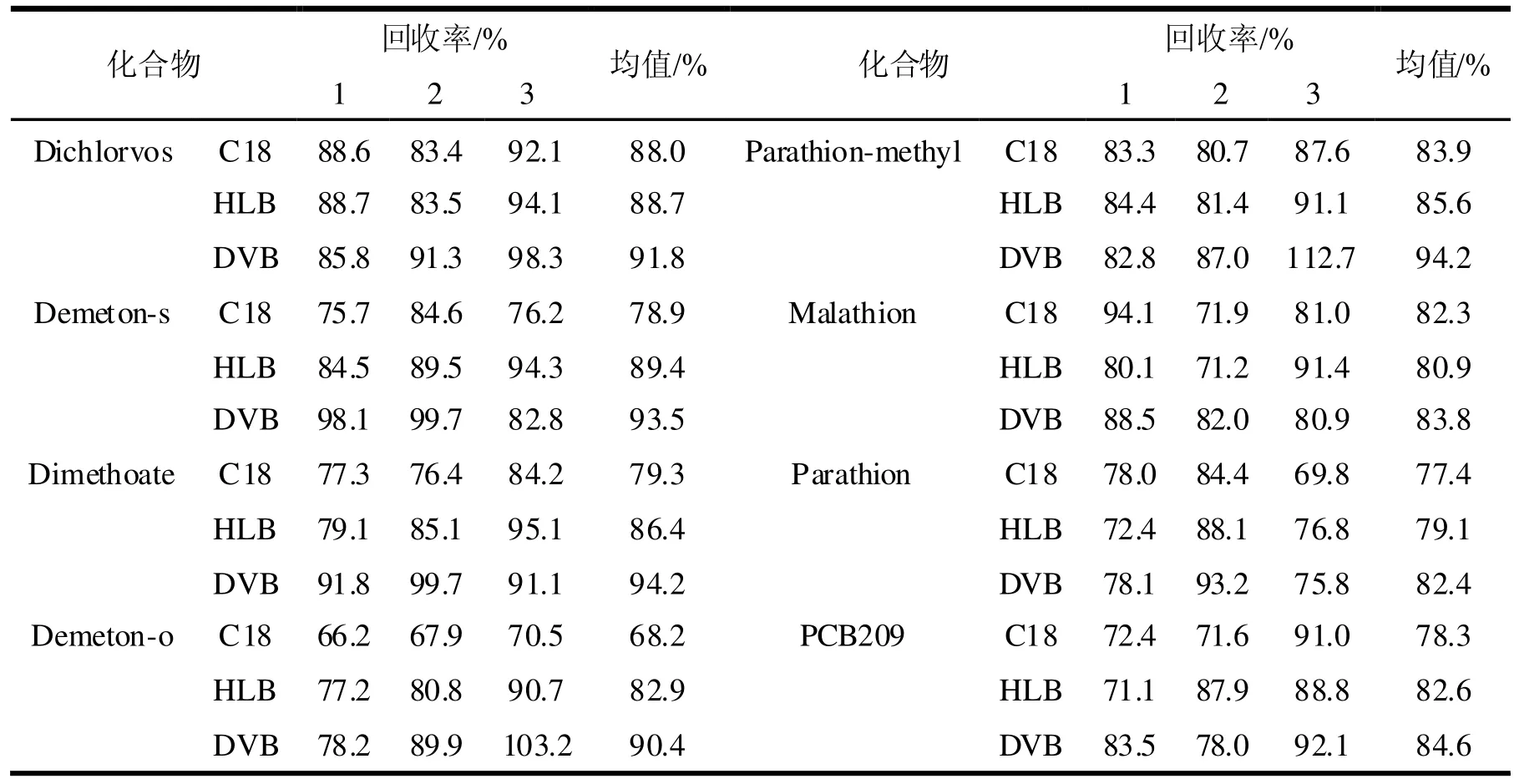
表2 不同萃取介質(zhì)的回收率
(2)萃取盤穿透性實驗
在分析低濃度樣品時,需要富集大體積的水樣,當樣品超過一定體積時,富集到固相萃取盤上的目標化合物會被洗脫一部分下來,導(dǎo)致回收率下降。對2 L (該萃取儀器的最大進樣體積) 0.001 mg/L的高濃度有機磷濃藥溶液進行萃取,將萃取后的溶液再進行萃取分析,均未發(fā)現(xiàn)目標化合物,因此本方法可富集至少2 L的樣品時目標化合物不被洗脫。
(3)萃取劑的選擇
常見的萃取劑有環(huán)已烷、正已烷、二氯甲烷、乙酸乙酯、丙酮和甲基叔丁基醚等,本文參美國EPA8141B[14]中所用萃取劑,即用丙酮和甲基叔丁基醚作為萃取劑。
(4)干吹時間的選擇
萃取盤是否吹干直接影響萃取效果,如果萃取盤沒吹干,水會因表面張力以及與有機溶劑極性的差導(dǎo)致萃取盤堵塞,使萃取效率降低,分用1、2、3、5、8、10 min試驗了干吹時間的影響,結(jié)果表明,當干吹時間少于3 min時,萃取盤容易被堵塞導(dǎo)致萃取不能進行,為了保證萃取盤被充分吹干以便獲得更好的萃取效率,實驗選用8 min為干吹時間。
(5)洗脫浸泡時間
試驗了洗脫浸泡時間對回收率的影響,由于horizon全自動固相萃取儀的洗脫液體積是不能優(yōu)化的,只能優(yōu)化浸泡時間,用0.0001 mg/L的有機磷溶液進行萃取,二氯甲烷和乙酸乙酯分別浸泡30、60、90、120 min后進行回收率測定。試驗發(fā)現(xiàn),60 min以后回收率達到最大,考慮到萃取效率和縮短萃取時間,本實驗選用丙酮浸泡90 min洗脫一次,MTBE浸泡90 min洗脫三次。
(6)脫水方式的選擇
樣品中如果有水,要損傷質(zhì)譜儀和降低質(zhì)譜儀的靈敏度,另外有水存在時,水與有機溶劑不互溶,當定容到1 mL時有機溶劑中目標化物的的濃度就不準確,因此在樣品處理過程中要除去萃取液中的水。傳
統(tǒng)的脫水方法用Na2SO4脫水,該方法要將Na2SO4在400℃下烘烤4 h,脫水前要用溶劑潤洗,脫水完后還要用有機溶劑沖洗,方法操作繁鎖,消耗試劑Na2SO4,同時也消耗了更多的有機溶劑,污染了環(huán)境,增加了氮吹濃縮時間,同時也增多了樣轉(zhuǎn)移次數(shù),從而降低了回收率。本實驗選用分子篩脫水,該分子篩用聚四氟乙烯作原料,做成一定大小的微孔,再用一些親有機物而疏水的材料涂于微孔之間,使有機物能通過微孔而水不能通過。方法操作簡單,樣品只轉(zhuǎn)移一次,不增加有機溶劑的量,脫水效果好,是一種環(huán)境友好型的前處理脫水方法。
(7)自動氮吹條件的選擇
自動氮吹儀的振蕩速度太快,溶劑揮發(fā)太快容易帶走少量目標化合物。振蕩速度太慢,氮吹時間加長,本實驗選振蕩速度為儀器最大值的60%,氮吹溫度的升高可使?jié)饪s速度加快,但隨溫度的升高回收率有所下降,本實驗選用30℃為氮吹溫度。
2.3標準曲線
用甲醇將有機磷農(nóng)藥和替代物PCB209配制成1.0 ug/mL的混合使用液,內(nèi)標物菲D10配制成1.0 ug/mL的使用液,分別取有機磷和替代物混合溶液5、10、20、50、100、200、500 uL用甲醇定容到1 mL再加入5 uL菲D10內(nèi)標溶液,則校準曲線的濃度分別為0.005、0.01、0.02、0.05、0.10、0.20、0.50 ug/mL,把校準曲線換算成500 mL水樣中工作曲線的質(zhì)量濃度分別為0.00001、0.00002、0.00004、0.0001、0.0002、0.0004、0.001 mg/L。用GC/MS選擇離子模式進行分析,內(nèi)標法進行定量,所得工作曲線、相關(guān)系數(shù)、線性范圍和檢出限見表3。由表3可見,方法檢出限在0.000006~0.000018 mg/L之間,相關(guān)系為0.9930~0.9991之間,表明方法有很好的靈敏度和線性關(guān)系。
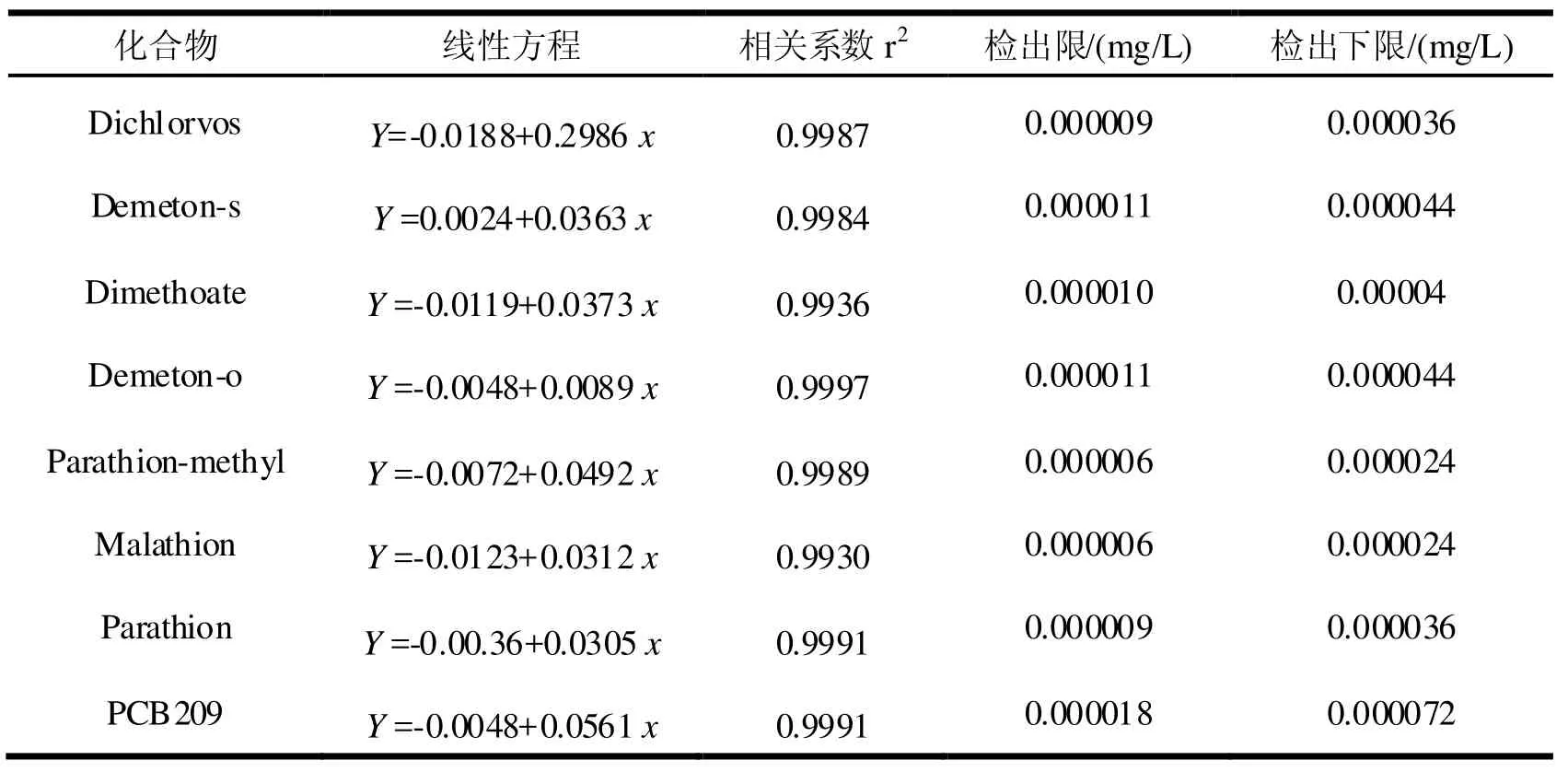
表3 標準曲線表
方法檢出限按《環(huán)境監(jiān)測分析方法標準制修訂技術(shù)導(dǎo)則》[15]進行測定,按照樣品分析步驟,對估計方法檢出限濃度的樣品進行7次平行測定,計算7次平行測定的標準偏差S,再按公式MDL(檢出限)=St (n-1, 0.99), t6, 0.99 =3.143計算檢出限,如果7次測定的平均值與MDL的比值在2~5之
間,則此MDL為該物質(zhì)方法檢出限,如果比值不在2~5范圍內(nèi),則增加或減少濃度重復(fù)以上步驟,直至比值在2~5范圍內(nèi),此時的結(jié)果為方法檢出限。
表4 精密度和加標回收率表(=6)
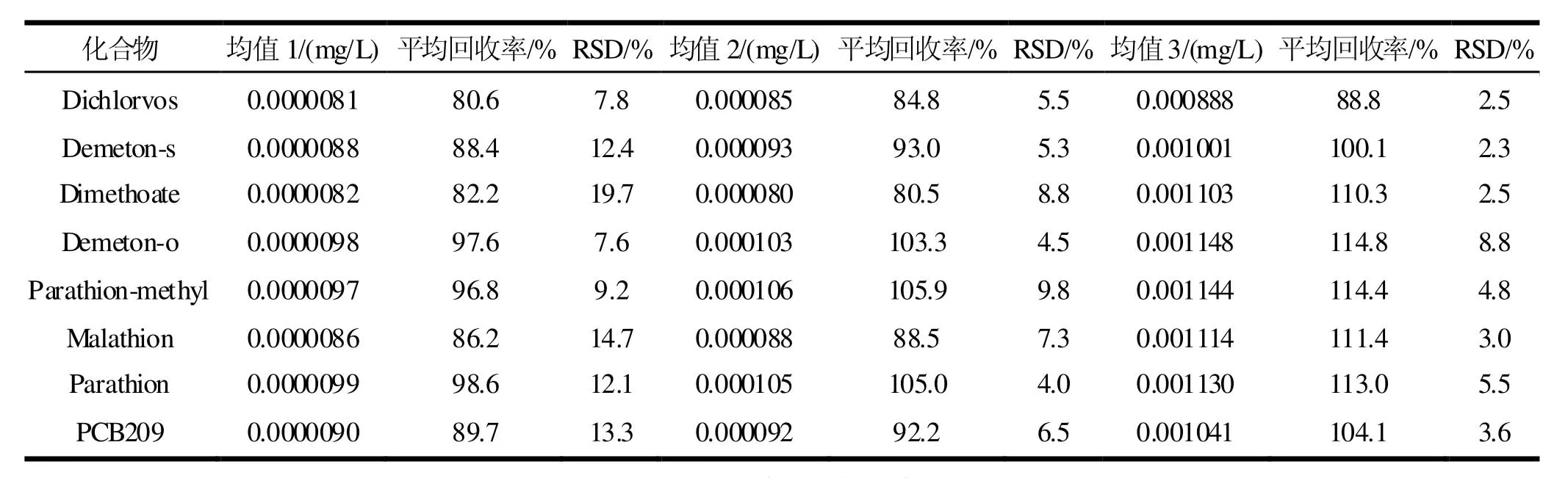
表4 精密度和加標回收率表(=6)
注:均值1、均值2、均值3的加標濃度分別為0.00001、0.0001、0.001 mg/L。
化合物 均值1 / ( m g / L ) 平均回收率/ % R S D / % 均值2 / ( m g / L ) 平均回收率/ % R S D / % 均值3 / ( m g / L ) 平均回收率/ % R S D / % D i c h l o r v o s 0 . 0 0 0 0 0 8 1 8 0 . 6 7 . 8 0 . 0 0 0 0 8 5 8 4 . 8 5 . 5 0 . 0 0 0 8 8 8 8 8 . 8 2 . 5 D e m e t o n -s 0 . 0 0 0 0 0 8 8 8 8 . 4 1 2 . 4 0 . 0 0 0 0 9 3 9 3 . 0 5 . 3 0 . 0 0 1 0 0 1 1 0 0 . 1 2 . 3 D i m e t h o a t e 0 . 0 0 0 0 0 8 2 8 2 . 2 1 9 . 7 0 . 0 0 0 0 8 0 8 0 . 5 8 . 8 0 . 0 0 1 1 0 3 1 1 0 . 3 2 . 5 D e m e t o n -o 0 . 0 0 0 0 0 9 8 9 7 . 6 7 . 6 0 . 0 0 0 1 0 3 1 0 3 . 3 4 . 5 0 . 0 0 1 1 4 8 1 1 4 . 8 8 . 8 P a r a t h i o n -m e t h y l 0 . 0 0 0 0 0 9 7 9 6 . 8 9 . 2 0 . 0 0 0 1 0 6 1 0 5 . 9 9 . 8 0 . 0 0 1 1 4 4 1 1 4 . 4 4 . 8 M a l a t h i o n 0 . 0 0 0 0 0 8 6 8 6 . 2 1 4 . 7 0 . 0 0 0 0 8 8 8 8 . 5 7 . 3 0 . 0 0 1 1 1 4 1 1 1 . 4 3 . 0 P a r a t h i o n 0 . 0 0 0 0 0 9 9 9 8 . 6 1 2 . 1 0 . 0 0 0 1 0 5 1 0 5 . 0 4 . 0 0 . 0 0 1 1 3 0 1 1 3 . 0 5 . 5 P C B 2 0 9 0 . 0 0 0 0 0 9 0 8 9 . 7 1 3 . 3 0 . 0 0 0 0 9 2 9 2 . 2 6 . 5 0 . 0 0 1 0 4 1 1 0 4 . 1 3 . 6
表5 實際樣品分析結(jié)果表(=6)

表5 實際樣品分析結(jié)果表(=6)
注:均值1、均值2、均值3分別是飲用水、工業(yè)廢水和生活污水樣品加標后有機磷的測定均值。
化合物 均值1 /(mg/L) 平均回收率/% RSD/% 均值2 /(mg/L) 平均回收率/% RSD/% 均值3 /(mg/L) 平均回收率/% RSD/% Dichlorvos 0.0000172 85.7 4.0 0.0000160 80.1 6.7 0.0000175 87.6 11.2 Demeton-s 0.0000184 92.2 6.5 0.0000169 84.6 8.7 0.0000182 91.0 11.2 Dimethoate 0.0000192 95.8 9.4 0.0000190 95.0 9.8 0.0000143 71.7 15.7 Demeton-o 0.0000181 90.7 7.5 0.0000184 91.8 11.1 0.0000202 101.2 8.5 Parathion-methyl 0.0000145 72.4 10.9 0.0000181 90.6 7.4 0.0000201 100.5 10.5 Malathion 0.0000187 93.1 7.8 0.0000159 79.4 16.4 0.0000162 80.8 16.7 Parathion 0.0000172 85.8 11.4 0.0000185 92.6 2.7 0.0000201 100.5 6.3
2.4精密度和準確度試驗
按《環(huán)境監(jiān)測分析方法標準制修訂技術(shù)導(dǎo)則》[15],用超純水做了高、中、低三個濃度級別的加標回收和精密度試驗,每種濃度各測定6次,試驗結(jié)果見表4。由表4可見,方法的加標回收率為80.6%~114.8%之間,相對標準偏差為2.3%~ 19.7%之間,表明方法準確可靠,能滿足分析要求。
3 樣品的測定
按實驗方法,對瀘州某飲用水斷面、某污水廠出水口污水和某化工廠污水處理設(shè)施出口廢水樣進行了測試和加標回收分析,每種樣品平行做兩個樣均未檢出目標化物,按《環(huán)境監(jiān)測分析方法標準制修訂技術(shù)導(dǎo)則》[15]通過基體加標使樣品中目標化物的濃度為0.00002 mg/L,平行分析6個加標樣,分析結(jié)果見表5。由表5可知,此法用于實際水樣中有機磷殘留的測定回收率在71.7%~101.2%之間,相對標準偏差在2.7% ~ 16.7%之間,表明方法準確可靠,滿足分析要求,可用于地表水、生活污水和工業(yè)廢水中有機磷農(nóng)藥殘留的測定。
4 結(jié)論
本文建立了全自動固相圓盤萃取,分子篩脫水技術(shù)對水中有機磷農(nóng)藥殘留進行提取分離,用GC/MS進行測定。方法自
動化程度高,精密度好,進樣量大,萃取時間少,溶劑用量少,回收率高,準確度高。方法使用分子篩脫水新技術(shù),操作簡單,樣品轉(zhuǎn)移次數(shù)少,不增加有機溶劑的用量,脫水效果好。整個前處理過程簡單快速,是一種綠色的環(huán)境友好前處理方法。方法用于地表水、工業(yè)廢水和生活污水中有機磷農(nóng)藥殘留的測定平均回率為71.7% ~ 101.2%,相對標準偏差為2.7% ~ 16.7%(=6),精密度和準確度均能滿足實際樣品分析要求。
參考文獻:
[1]Khan I A,Riazuddin,Parveen Z,Multi-residue det ermination of synthetic pyrethroids and organophosphorus pesticides in whole wheat flour using gas chromatography [J].Bull Environ ContamToxicol,2007,79 (4): 454-460.
[2]Ballesteros E,Parrado M J,Continuous solid-phase extraction and gas chromatographicdeterminationof organophosphorus pesticides in natural and drinking water [J]. Journal of ChromatographyA,2004,1029 (1-2):267-273.
[3]Khalili-Zanjani MR,Yamini Y,Yazdanfar N,Extraction and determination of organophosphorus pesticides in waters amples by a new liquid phase micro extraction-gas chromatography-flame photometric detection[J].Analytica chimica acta,2008,606 (2): 202-208.
[4]Yao Z W,Gui B J,Jie M L,Application of solid-phase microextraction for the determination of organophosphorous pesticides in aqueous samples by gas chromatography with flame photometricdetector [J].Talanta,2001,55 : 807-814.
[5]Christos J P.Nikolaos V K.A comparison of dimethoate degradation in lemons and mandarins on the trees with two GCsystems [J].Food Chemistry,2003,80 : 23-28.
[6]John G B,Paul R H,Gerard J S.Desmarchelierdeterminationof organophosphate pesticides and carbaryl on paddy rice by reversed-phase high-performance liquid chromatography [J].Journal of Chromatography,1988,447 (1): 249-255.
[8]Khuhawar M Y,Rind F W A,Almani K F,Spectrophotometric determination of tranexamic acid in dosage forms by derivatization[J].Jour Chem Soc Pak,2006,28(5): 435-438.
[9]Raina R,Sun L.Trace level determination of selected organophosphorus pesticides and their degradation products in environmental air samples by liquid chromatography-positive ion electrospray tandem mass spectrometry [J].Journal of environmental science and health part B-pesticides food contaminants and agricultural wastes,2008,43(4): 323-332.
[10]Norman K N,Panton S H.Supercritical fluid extraction and quantitative determination of organophosphorus pesticide residues in wheat and maize using gas chromatography with flame photometric and mass spectrometric detection [J].Journal of Chromatography,2001,907 (1-2 ): 247-258.
[11]Schellin M,Hauser B,Popp P.Determination of organophosphorus pesticides using membrane-assisted solvent extraction combined with large volume injection-gas chromatography-mass spectrometric detection[J].Journal of Chromatography A,2004,1040 (2): 251-258.
[12]Vidal J L M,Arrebola F J,Mateu-SanchezM.Multi-residue method for determination of pesticides invegetablesamples by GC-MS-MS.Chromatographia,2002,56(7/8): 475.
[13]Blasco C,F(xiàn)ernandez M,Pico Y,Comparison of solid-phase micro-extractionandstirbarsorptive extractionfordeterminingsix organophosphorusinsecticidesin honeybyliquidchromatography-mass spectrometry [J].Journal of chromatography A,2004,1030 (1-2): 77-85.
[14]EPA METHOD 8141-1994 A, organophosphorus compounds by gas chromatographycapillarycolumn technique [S].
[15]HJ168-2010,Environmental monitoring-Technical guideline on drawing and revising analytical method[S] (in Chinese).
Using Automated Solid Phase Extractionand Molecular Sieve Dehydration Coupled with Gas Chromatography/Mass Spectrometry
QIN Ming-you
(Jinshan Environmental Protection Monitoring Station, Shanghai 200540,China)
Abstract:Automated solid phase extraction and molecular sieve dehydration coupled with gas chromatog-raphy/mass spectrometry (GC/MS) was used to determine the residues of organophosphorous pesticides in water samples.The extraction disk penetrating was tested and the extraction condition was optimized. Using acetone and methyl tert-butyl ether(MTBE)as the eluen and DVB disk as the extraction disk, the pro-posed method has been successfully applied for the determination of organophosphorous pesticides residues in drinking water, industrial wastewater and raw sewage with satisfactory results for its sensitivity, precision and repeatability. The average recoveries were between 71.7%-101.2% with RSD of 2.7%-16.7%(n=6). The limits of detection was between 0.000006~0.000018 mg/L.
Key words:organophosphorous pesticides; molecular sieve dehydration; automated solid Phase extraction; gas chromatography/Mass spectrometry; water
作者簡介:第一秦明友(1975-),男,四川瀘縣人,博士,高級工程師,主要從事環(huán)境監(jiān)測和儀器分析工作。
收稿日期:2014原08原05
中圖分類號院韻661.1
文獻標志碼院A
文章編號:1671-1602(2015)08-0037-07
猜你喜歡
中國纖檢(2016年12期)2017-01-20 09:28:19
現(xiàn)代農(nóng)業(yè)科技(2016年20期)2016-12-20 14:51:09
現(xiàn)代農(nóng)業(yè)科技(2016年20期)2016-12-20 09:05:36
分析化學(xué)(2016年7期)2016-12-08 00:09:44
分析化學(xué)(2016年7期)2016-12-08 00:07:08
價值工程(2016年29期)2016-11-14 01:34:54
科技視界(2016年24期)2016-10-11 18:58:00
考試周刊(2016年39期)2016-06-12 16:01:44
中國科技博覽(2016年4期)2016-04-25 07:25:47
中國科技博覽(2016年8期)2016-04-25 04:57:50
