廢MTP催化劑對模擬廢水中對二甲苯的吸附作用
2020-09-16 07:25:24趙娜娜金政偉張安貴
凈水技術 2020年9期
趙娜娜,金政偉,莊 壯,張安貴,蘇 慧
(國家能源集團寧夏煤業有限責任公司煤炭化學工業技術研究院,寧夏銀川 750411)
對二甲苯[1]是一類普遍使用的化工原料,也是許多工業企業生產的副產物。根據《全球化學品統一分類和標簽制度》(GHS),PX為易燃液體,會造成水污染,蒸氣接觸會導致中毒:對皮膚和眼的刺激性和全身(神經中樞)毒性;據《職業性接觸毒物危害程度分級》(GB 5044—B 85),PX為中度危害級別物質,與鹽酸、甲醇屬于同一級別;根據《石油化工企業設計防火規范》(GB 50160—2008),PX屬于甲B類,火災危險性低于液化烴及與其類似液體[2]。苯系物的工業污染源主要為石油化工生產的排放廢水。隨著鋼鐵冶金、焦化、煉油、石化等工業的飛速發展,含二甲苯廢水的種類和數量日益增多。這種廢水不僅危害農業生產、動植物生長繁殖,甚至還會威脅人類健康。目前,處理對二甲苯的主要技術有熔融結晶技術、精餾技術、膜分離技術及吸附分離技術。精餾技術和熔融結晶技術缺點在于能量耗費巨大且分離產品純度很低,效率低下,而膜分離技術限制于膜材料批量生產困難和使用壽命較短[1,3]。基于此,吸附技術因投資和生產成本低且選擇性較高成為含油污水適度處理的常用技術之一,吸附法所用吸附劑的選擇是該技術的核心[4-6]。利用多孔介質較大的比表面積和豐富的孔道結構,可以選擇性地吸附去除有機污染物[7-8]。分子篩吸附劑[9-11]在工業上的應用發展很快,與活性炭吸附劑相比,具有脫除率高、凈化徹底、能耗低、再生性能好等優點,被廣泛應用于工業廢水中有機物的凈化處理[12-14]。
廢催化劑是固體廢棄物的主要來源[15]。由于環境問題日益嚴峻,越來越多的學者將重點放在廢催化劑盡可能地資源化利用過程上。國家能源集團寧夏煤業有限責任公司50萬t/a煤基聚丙烯項目甲醇制丙烯(MTP)技術采用德國魯奇工藝。MTP催化劑在固定床反應器內使用壽命為7 000~8 000 h,下線的MTP催化劑經過簡單的器內再生后,直接卸除作為“危廢”處理,每年產生廢MTP催化劑約450 t[16]。經表征分析發現,該廢催化劑中活性組分骨架結構仍保持良好,且具有比表面積大、孔隙率高、吸附性能良好等特點,若經過適度活化及成型等工藝處理,應用到有機廢水的凈化過程,既能實現廢催化劑的資源化利用,又可大大降低有機廢水處理成本。本文采用酸堿硅烷化改性廢MTP催化劑作為疏水吸附劑來處理含對二甲苯的模擬廢水,通過加入酸或堿脫除分子篩的鋁原子或硅原子來改變分子篩的硅鋁比,加入硅烷化試劑進一步提高疏水性,由此獲得適宜的工藝條件,取得了較為滿意的效果,可為廢催化劑處理有機廢水的研究提供一定的理論基礎和數據支持。
1 材料和方法
1.1 試驗儀器和試劑
試劑:甲苯(南京合誠化工有限公司,AR),對二甲苯(PX)(成都艾科達化學試劑有限公司,GR),草酸(Aladdin,AR),68%HNO3(成都市科隆化學品有限公司,AR),NaOH(天津市北聯精細化學品開發有限公司,AR),正辛基三乙氧基硅烷(OTS)(Aladdin,97%),廢MTP催化劑(國家能源集團寧夏煤業有限責任公司烯烴一分公司MTP反應器取出),本試驗所用水均為去離子水。
儀器:氣相色譜儀(Agilent 6820,HP-PLOT/Q毛細柱)。
1.2 分析方法
采用美國安捷倫Agilent 6820型氣相色譜測定對二甲苯含量。采用HP-PLOT/Q柱,30 m0-PLO mm-PLOT μm;升溫程序:初始溫度為35 ℃,以2 ℃/min升溫至200 ℃保持20 min;載氣流速為1.2 mL/min的高純氮氣(>99. 999%);分流比為2∶1。進樣口溫度為220 ℃。
采用荷蘭帕納科X’pert3 powder型X-射線粉末衍射儀(XRD)測定改性前后廢MTP催化劑的晶體物相結構,工作電壓為40 kV,管電流為40 mA,銅靶,1維半導體PIXcel1D矩陣探測器,掃描方式為 θ/θ,最小步長為0.0001°,掃描范圍為5°~90°。
采用德國布魯克公司的BRUKER V70紅外光譜儀表征改性前后廢MTP催化劑的內部骨架結構,KBr壓片,波數為400~4 000 cm-1。
1.3 試驗方法
稱取一定量廢MTP催化劑過篩,取200目以下的細粉于管式爐空氣氣氛下600 ℃焙燒4~6 h,冷卻至室溫。采用4 mol/L草酸于95 ℃、液固比為(5~10)∶1下處理廢催化劑4 h,離心分離(5 000 r/min,3 min),去離子水洗滌,鼓風干燥箱中120 ℃過夜干燥,酸處理后的廢催化劑加入到0.2 mol/L NaOH溶液(固液比為10∶1)中,在95 ℃水浴中機械攪拌處理2 h,離心分離(5 000 r/min,3 min),將離心得到的上層廢催化劑泥漿再用1 mol/L稀HNO3溶液攪拌洗滌30 min以脫除處理后產生的殘留物,去離子水洗滌至中性后,置于鼓風干燥箱中120 ℃過夜干燥,馬弗爐600 ℃焙燒6 h,得到酸堿處理產品。
稱取一定量酸堿處理產品加入正辛基三乙氧基硅烷(OTS)試劑(固液比為5∶1)置于500 mL晶化釜中,120 ℃反應10 h,冷卻后離心分離,無水甲醇洗滌4~6次,70 ℃真空干燥過夜,得到吸附劑產品,測其XRD譜圖,如圖1所示。由圖1可知,由廢MTP催化劑制備的吸附劑產品保持著典型的ZSM-5分子篩結構,在2θ為7.9°、8.8°、23.1°和23.8°處出現分別對應ZSM-5(011)、(020)、(051)和(033)晶面的特征峰,其結晶度較高,說明分子篩孔道長程有序。
吸附劑產品的紅外譜圖如圖2所示。由圖2可知,使用正辛基三乙氧基硅烷試劑改性后,ZSM-5骨架峰未發生明顯變化,表明分子篩的骨架結構基本不發生變化。1 107 cm-1和804 cm-1處吸收峰歸屬于硅氧四面體內部不對稱伸縮振動峰,457 cm-1處吸收峰為Si-O-Si彎曲振動,1 230 cm-1處吸收峰歸屬于ZSM-5骨架內硅氧四面體和鋁氧四面體不對稱伸縮振動峰[17]。在2 859、2 928、2 960 cm-1處出現了-CH3、-CH2的吸收峰,這表明OTS改性,-CH3、-CH2取代了ZSM-5結構中的Si-OH,-Si(CH2)7CH3基團成功接枝在廢MTP催化劑表面,提高了吸附劑的疏水性。
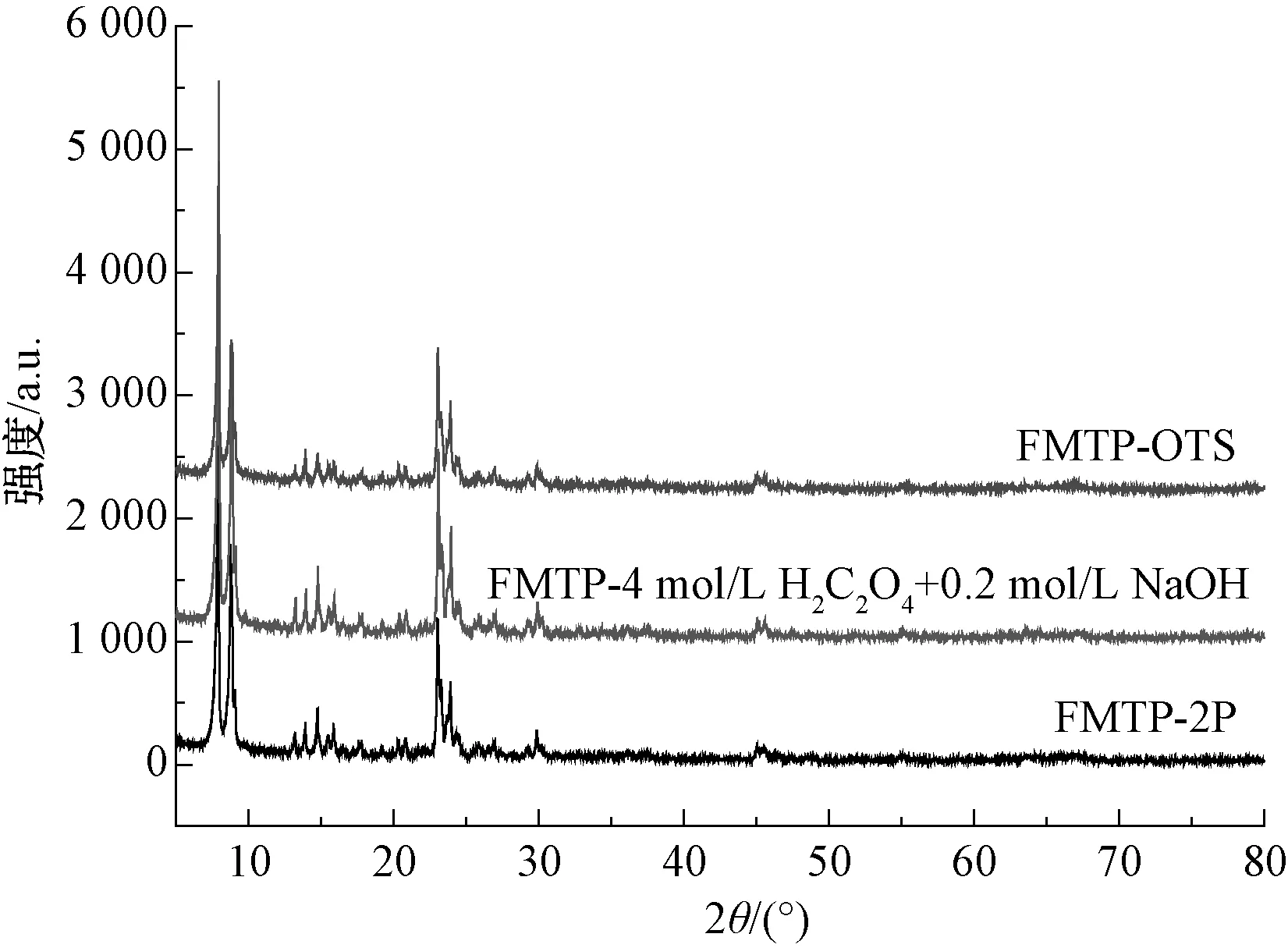
圖1 吸附劑產品的XRD譜圖Fig.1 XRD Patterns of Adsorbent Products

圖2 吸附劑產品的IR譜圖Fig.2 FT-IR Patterns of Adsorbent Products
1.4 PX標準曲線繪制
在6個25 mL的錐形瓶中,分別加入不同濃度的PX,再加入20 g甲苯,配制成0.02%~1.2%的PX-甲苯溶液,充分搖動溶液,吸取1 mL溶液在氣相色譜上測其峰高,繪制PX標準曲線[18],如圖3所示。

圖3 PX標準曲線Fig.3 Standard Curve of PX
1.5 吸附試驗
在一系列250 mL碘量瓶中分別加入不同質量的吸附劑和100 mL不同濃度的對二甲苯水溶液,調節pH后,在不同溫度下,置于磁力攪拌器攪拌一定時間至吸附平衡,離心分離,加入一定量的甲苯,震蕩搖勻,靜置,吸取上層溶液,在氣相色譜儀上測定PX的含量。在5種不同的條件下,當吸附率即PX濃度不變時,證明吸附已達平衡[19]。
通過式(1)計算PX的吸附率(X)[19]。
(1)
其中:C0——起始PX質量濃度,mg/L;
C1——吸附平衡后PX質量濃度,mg/L。
通過式(2)計算PX的吸附量[19]。
(2)
其中:qt——t時刻廢MTP催化劑的吸附量,mg/g;
C0——起始PX質量濃度,mg/L;
Ct——吸附t時刻后PX質量濃度,mg/L;
m——廢MTP催化劑質量,g;
V——液相總體積,mL。
1.6 廢MTP催化劑的吸附動力學分析和吸附等溫線分析
1.6.1 吸附速率方程公式
本試驗一級速率常數通過Lagergren-Svenska一級速率方程[20]計算,如式(3)。
lg(qe-qt)=lgqe-K1t
(3)
其中:qe——吸附劑平衡時刻的吸附量,mg/g;
qt——吸附劑t時刻的吸附量,mg/g;
K1——吸附速率常數,L/min;
t——吸附時間,min。
二級吸附速率方程[20]如式(4)。
(4)
其中:qe——吸附劑平衡時刻的吸附量,mg/g;
K2——吸附速率常數,L/min;
t——吸附時間,min。
1.6.2 吸附等溫線
Langmuir等溫吸附方程[21]如式(5)所示。
(5)
其中:qe——飽和吸附量,mg/g;
qm——最大吸附量,mg/g;
Ce——平衡濃度,mg/L;
Kl——常數。
Freundlich等溫吸附方程[21]如式(6)所示。
(6)
其中:Kf——常數;
n——常數。
2 結果與討論
2.1 各吸附條件對廢MTP催化劑吸附性能的影響
2.1.1 吸附時間對吸附性能的影響
圖4為吸附率隨吸附時間變化的關系。由圖4可知:當吸附時間為20 min時,吸附率為88%;隨著吸附時間的增長,改性廢MTP催化劑對PX的吸附非常快;當吸附時間為60 min時,PX的吸附率達到94.3%;此后,繼續延長時間,吸附速率變化不大;當吸附時間為120 min時,吸附率為94.0%。這是由于廢水中初始PX濃度大,改性廢MTP催化劑吸附點位周圍有很高濃度的PX,吸附速率大于解析速率,開始時吸附率增大,當吸附時間為60 min時,吸附已平衡,廢水中PX濃度變化不大,此時吸附達到飽和狀態[22]。表明,廢催化劑可以迅速、高效地吸收廢水中的對二甲苯。
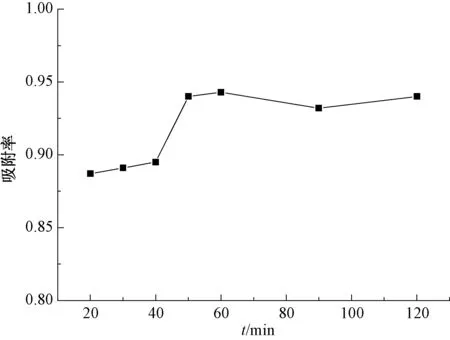
圖4 吸附時間對PX吸附率的影響Fig.4 Effect of Different Absorption Time on Adsorption Rate of PX
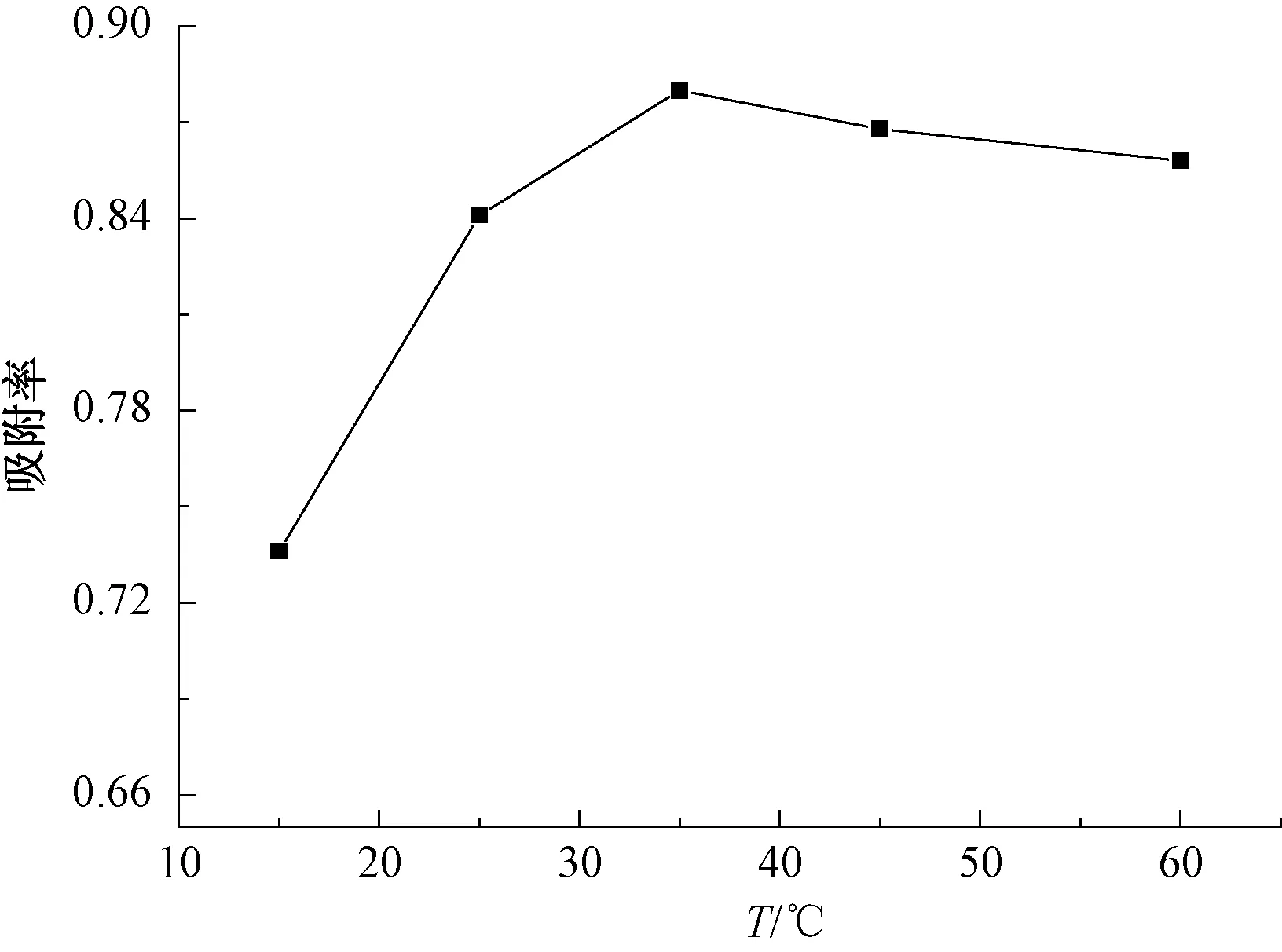
圖5 吸附溫度對PX吸附率的影響Fig.5 Effect of Different Absorption Temperature on Adsorption Rate of PX
2.1.2 溫度對吸附性能的影響
圖5為吸附率隨吸附溫度變化的關系。由圖5可知:改性廢MTP催化劑對PX的吸附率隨溫度的增加而增加;在35 ℃時,吸附效果最好,吸附率達到88%;繼續升高溫度,吸附率下降。原因為溫度升高,體系中各基團活性上升,導致改性廢MTP催化劑吸附對二甲苯的速率上升,吸附率升高。同時,吸附過程是可逆的,溫度升高使得對二甲苯脫附速率加快,吸附率降低,這與文獻報道一致[23]。改性廢MTP催化劑吸附PX的最佳溫度為35 ℃。
2.1.3 吸附劑量對吸附性能的影響
圖6為吸附率隨吸附劑量變化的關系。由圖6可知:PX的吸附率隨吸附劑量的增加而提高;當改性廢MTP催化劑用量達到1.0 g時,水中對二甲苯的吸附率達到83%;此后,隨吸附劑量的增加,吸附率卻有所降低。這是由于溶液濃度一定時,吸附劑用量少,吸附的活性位點少,PX的吸附率也較低,隨著吸附劑量的增多,吸附活性位增加,吸附率逐漸增大。但是,當吸附劑量為1.0 g時,吸附活性位趨于飽和,能夠對420 mg/L的PX進行很好的吸附,幾乎已經達到吸附平衡,再繼續增加吸附劑用量,對吸附效果影響不大;或吸附較多時,吸附劑本身會顆粒黏附,碰撞概率加大[24-25],發生抑制吸附,使吸附效果下降[26-27]。
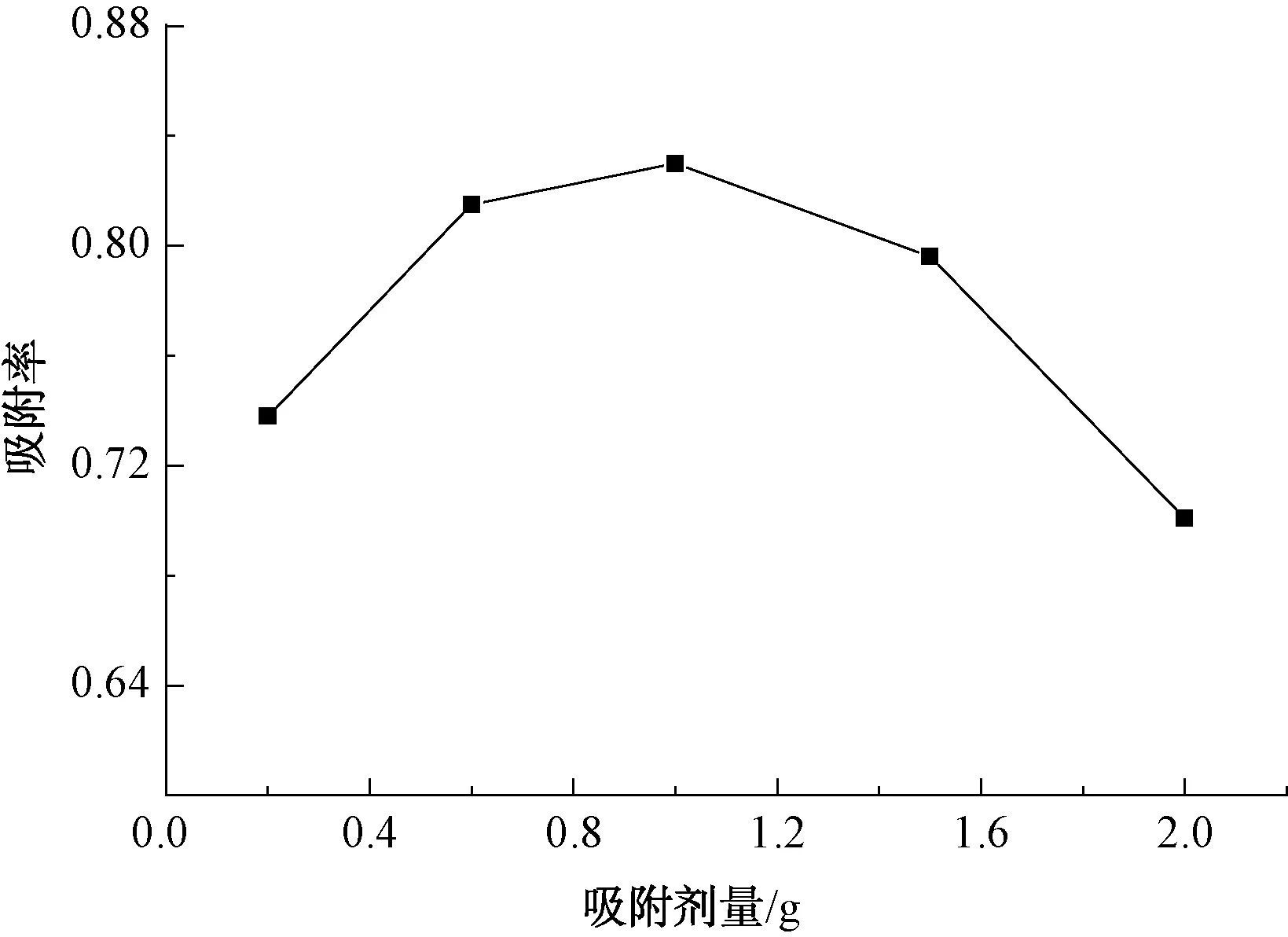
圖6 吸附劑量對PX吸附率的影響Fig.6 Effect of Different Absorption Dosage on Adsorption Rate of PX
2.1.4 pH值對吸附性能的影響
圖7是吸附率隨pH變化的關系。由圖7可知,改性廢MTP催化劑在酸性及中性條件下對PX的吸附效果明顯好于堿性條件下的吸附效果。吸附率最大對應的溶液pH值為5.0。當pH增加時,溶液中的OH-增加。PX與OH-發生競爭吸附,從而影響PX的吸附。而當pH降低時,溶液中的H+增加,抑制OH-的產生,從而促進吸附的進行[28]。但酸性過高,廢MTP催化劑的吸附位點可能被多余的H+占據,吸附PX的量反而減少,使得吸附率降低。
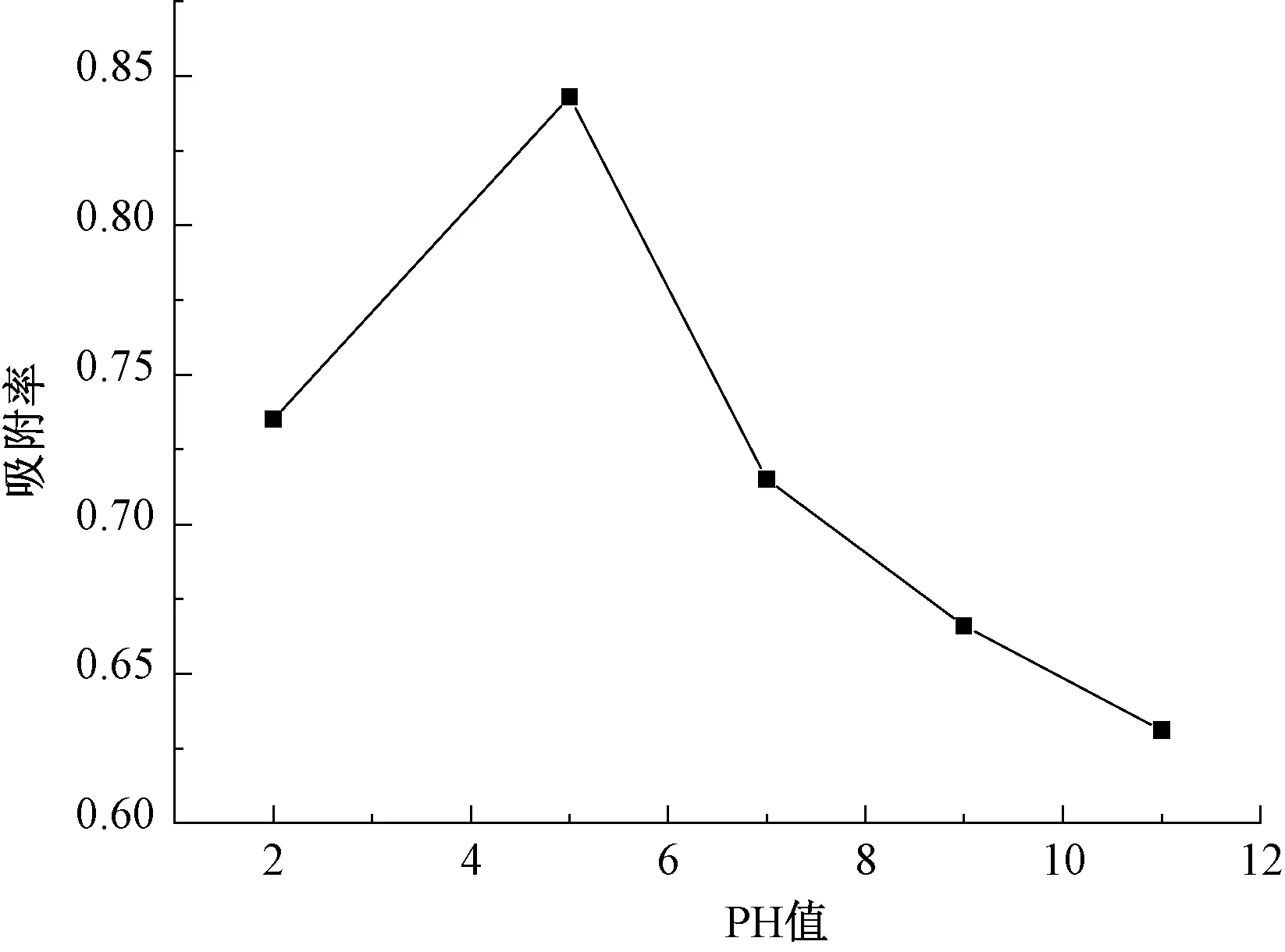
圖7 pH對PX吸附率的影響Fig.7 Effect of Different pH Value on Adsorption Rate of PX
2.1.5 PX廢水初始濃度對吸附性能的影響
圖8是吸附率隨PX初始濃度變化的關系。由圖8可知,隨著PX廢水初始濃度的增大,吸附率逐漸增大。當初始濃度為1.3 g/L時,吸附達到飽和。原因是PX濃度較低時,吸附劑提供的活性位點數量遠大于 PX的數量,故隨著PX濃度的增大,改性廢MTP催化劑對PX的吸附也越大。當PX濃度超過1.3 g/L時,此時活性位點數少于PX的被吸附量,出現競爭吸附[29]。總體而言,最佳吸附率為88.0%。
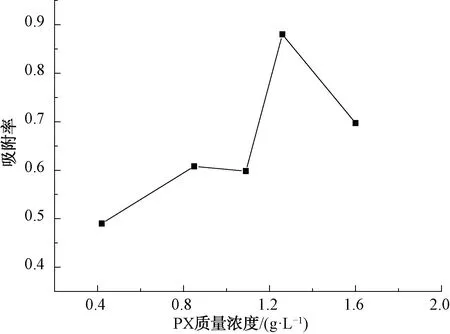
圖8 PX初始濃度對PX吸附率的影響Fig.8 Effect of Different Initial PX Concentration on Adsorption Rate of PX
由圖8可知,改性廢MTP催化劑吸附對二甲苯廢水的影響因素主要有吸附時間、溫度、吸附劑量、pH以及PX廢水初始濃度。由此,設計正交試驗,如表1所示。
正交試驗所得的結果如表2所示。
對結果進行極差分析,計算4個水平吸附率的平均值和極差R,如表3所示。
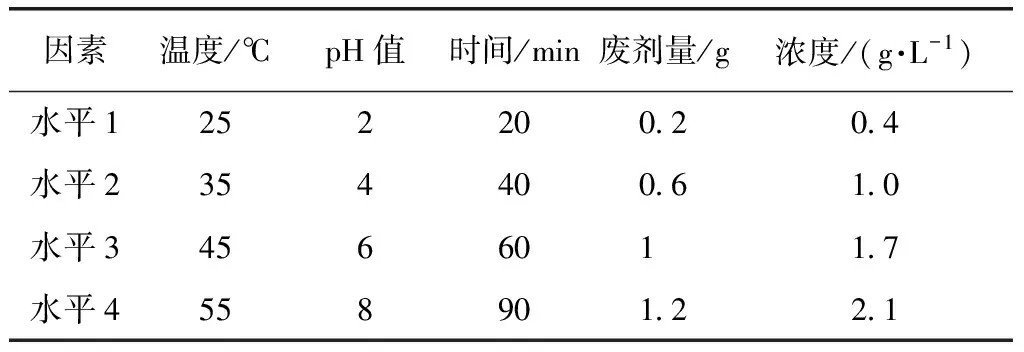
表1 正交試驗因素水平Tab.1 Factor Levels of Orthogonal Experiments
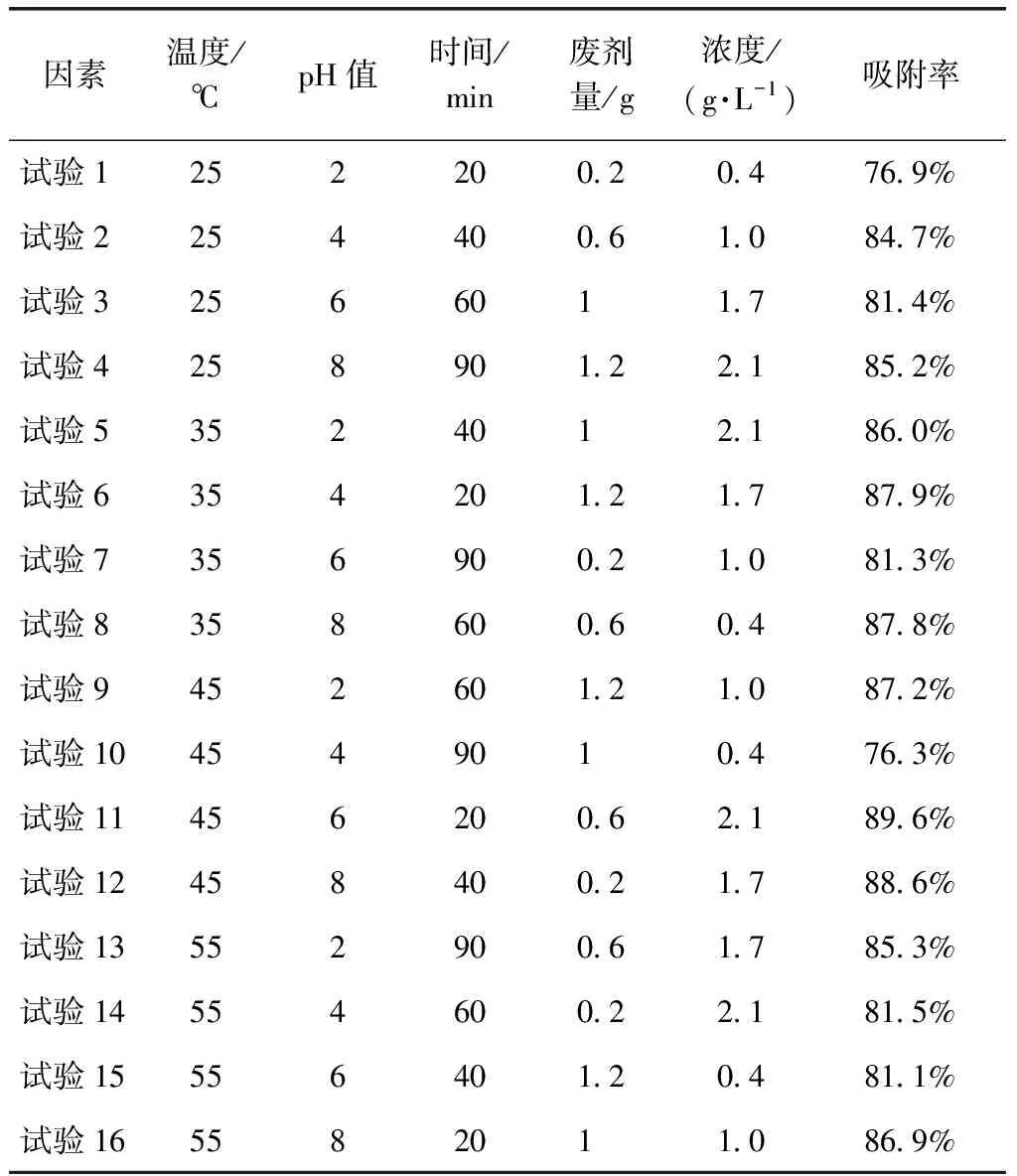
表2 正交試驗結果Tab.2 Results of Orthogonal Experiment
由表3可知,在以上5個因素中,影響吸附率的主要因素依次為:PX廢水初始濃度>廢催化劑用量>pH>溫度>時間。由正交試驗結果可知,改性廢MTP催化劑處理廢水中PX的最佳條件:溫度為45 ℃,pH值為6,吸附時間為20 min,改性廢MTP催化劑用量為0.6 g,PX廢水初始濃度為2.1 g/L。
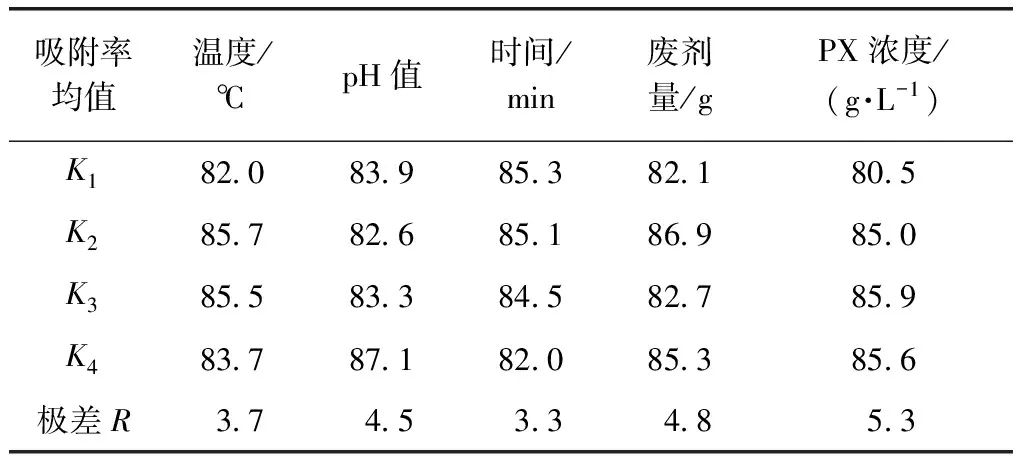
表3 極差分析結果Tab.3 Results of Range Analysis
2.2 廢MTP催化劑的吸附動力學分析和吸附等溫線分析
2.2.1 廢MTP催化劑的吸附動力學分析
根據式(3)~式(5)計算得表4。
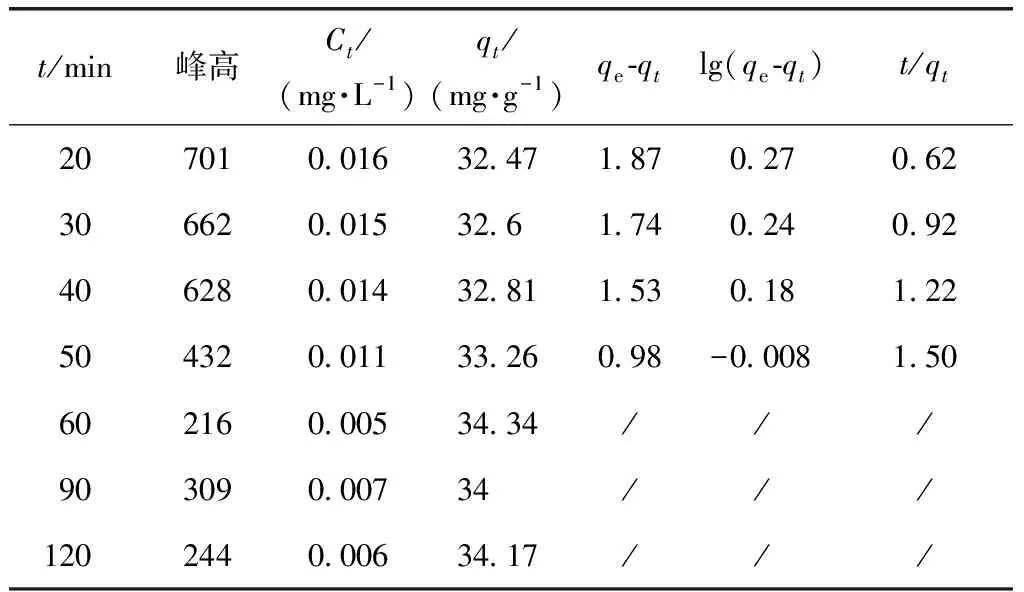
表4 廢MTP催化劑的吸附動力學分析Tab.4 Adsorption Kinetics Analysis of Spent MTP Catalyst
圖9為廢MTP催化劑的一級速率方程回歸曲線和二級速率回歸曲線。
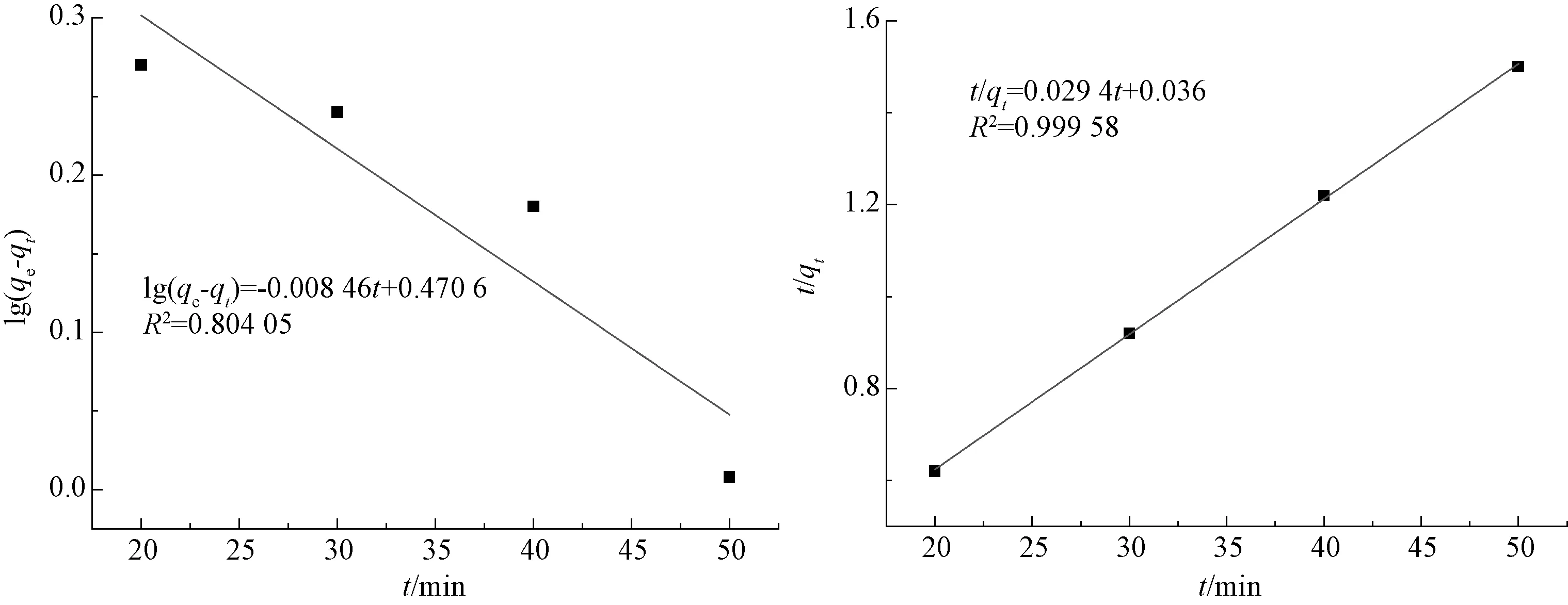
圖9 一級速率方程回歸曲線圖(左)和二級速率方程回歸曲線圖(右)Fig.9 Regression Curve of First-Order Rate Equation (Left) and Regression Curve of Second-Order Kinetic Equation (Right)
由圖9可知,一級速率方程的線性相關系數R2=0.804 05,二級速率方程的線性相關系數R2=0.999 58。由于二級速率方程回歸時擬合效果較一級速率方程回歸時好,本吸附試驗符合二級吸附動力學方程。
2.2.2 廢MTP催化劑的吸附等溫線分析
廢MTP催化劑的吸附等溫線數據如表5所示。
廢MTP催化劑的Langmuir吸附等溫線回歸曲線和Freundlich吸附等溫線回歸曲線如圖10所示。
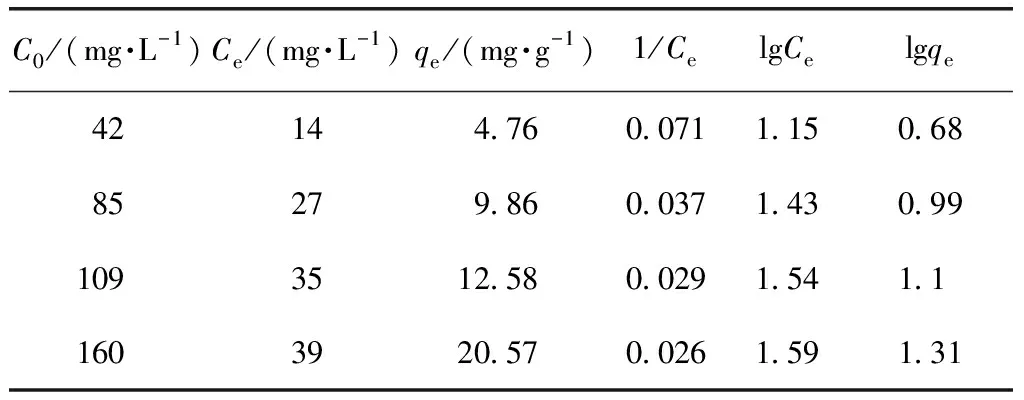
表5 廢MTP催化劑的吸附等溫線數據Tab.5 Adsorption Isotherm Data of Spent MTP Catalyst

圖10 Langmuir等溫線回歸圖(左)和Freundlich等溫線回歸圖(右)Fig.10 Regression Curve of Langmuir Isotherm (Left) and Regression Curve of Freundlich Isotherm (Right)
由圖10可知,Langmuir吸附等溫線線性相關系數R2=0.977 83,Freundlich吸附等溫線線性相關系數R2=0.912 14。Langmuir吸附等溫線對PX在廢MTP催化劑上的吸附數據做了更為準確的解析,本吸附試驗符合Langmuir吸附等溫線。
2.3 廢MTP催化劑循環利用性能
選用2.1 g/L的PX模擬廢水進行吸附試驗。將已經吸附飽和的PX-改性廢MTP催化劑溶液常壓過濾,140 ℃焙燒6 h后用于吸附-焙燒解吸-再吸附試驗,考察經多次循環使用后PX的平衡吸附量變化,如圖11所示。

圖11 廢MTP催化劑吸附PX平衡吸附圖Fig.11 Equilibrium Adsorption Diagram of Spent MTP Catalyst Circulating Adsorption of PX
由圖11可知,吸附平衡后的改性廢MTP催化劑經焙燒解吸,在相同情況下對PX的吸附量基本不變。說明,該吸附劑的耐損性能較好,可再生并多次循環使用。
3 結論
本研究選用酸堿硅烷化改性廢MTP催化劑作為吸附劑,吸附模擬廢水中的對二甲苯,得出如下結論。
(1)改性廢MTP催化劑可以迅速、高效地吸收廢水中的對二甲苯。通過考察吸附溫度和吸附時間等因素對PX的影響規律,得到其最佳條件:吸附溫度為45 ℃,pH值為6,吸附時間為20 min,酸改性廢MTP催化劑用量為0.6 g,PX廢水初始濃度為2.1 g/L。此時,PX的吸附率最高達到了89.6%。
(2)借助XRD表征酸堿硅烷化改性后廢MTP催化劑物相,結果表明,其仍保持著典型的ZSM-5分子篩結構。IR發現,改性后骨架結構基本不發生變化,-CH3,-CH2取代了ZSM-5結構中的Si-OH。使用正辛基三乙氧基硅烷改性可成功將-Si(CH2)7CH3基團接枝在廢MTP催化劑表面,提高了吸附劑的疏水性。
(3)廢MTP催化劑吸附廢水中的對二甲苯符合二級吸附動力學,廢MTP催化劑對PX的吸附符合Langmuir等溫吸附。
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
中國塑料(2016年5期)2016-04-16 05:25:36
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
